 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSimilarity-Quantized Relative Difference Learning for Improved Molecular Activity Prediction
Jan 15, 2025



Accurate prediction of molecular activities is crucial for efficient drug discovery, yet remains challenging due to limited and noisy datasets. We introduce Similarity-Quantized Relative Learning (SQRL), a learning framework that reformulates molecular activity prediction as relative difference learning between structurally similar pairs of compounds. SQRL uses precomputed molecular similarities to enhance training of graph neural networks and other architectures, and significantly improves accuracy and generalization in low-data regimes common in drug discovery. We demonstrate its broad applicability and real-world potential through benchmarking on public datasets as well as proprietary industry data. Our findings demonstrate that leveraging similarity-aware relative differences provides an effective paradigm for molecular activity prediction.
Efficient Fine-Tuning of Single-Cell Foundation Models Enables Zero-Shot Molecular Perturbation Prediction
Dec 18, 2024



Predicting transcriptional responses to novel drugs provides a unique opportunity to accelerate biomedical research and advance drug discovery efforts. However, the inherent complexity and high dimensionality of cellular responses, combined with the extremely limited available experimental data, makes the task challenging. In this study, we leverage single-cell foundation models (FMs) pre-trained on tens of millions of single cells, encompassing multiple cell types, states, and disease annotations, to address molecular perturbation prediction. We introduce a drug-conditional adapter that allows efficient fine-tuning by training less than 1% of the original foundation model, thus enabling molecular conditioning while preserving the rich biological representation learned during pre-training. The proposed strategy allows not only the prediction of cellular responses to novel drugs, but also the zero-shot generalization to unseen cell lines. We establish a robust evaluation framework to assess model performance across different generalization tasks, demonstrating state-of-the-art results across all settings, with significant improvements in the few-shot and zero-shot generalization to new cell lines compared to existing baselines.
RINGER: Rapid Conformer Generation for Macrocycles with Sequence-Conditioned Internal Coordinate Diffusion
May 30, 2023



Macrocyclic peptides are an emerging therapeutic modality, yet computational approaches for accurately sampling their diverse 3D ensembles remain challenging due to their conformational diversity and geometric constraints. Here, we introduce RINGER, a diffusion-based transformer model for sequence-conditioned generation of macrocycle structures based on internal coordinates. RINGER provides fast backbone sampling while respecting key structural invariances of cyclic peptides. Through extensive benchmarking and analysis against gold-standard conformer ensembles of cyclic peptides generated with metadynamics, we demonstrate how RINGER generates both high-quality and diverse geometries at a fraction of the computational cost. Our work lays the foundation for improved sampling of cyclic geometries and the development of geometric learning methods for peptides.
CREMP: Conformer-Rotamer Ensembles of Macrocyclic Peptides for Machine Learning
May 14, 2023Computational and machine learning approaches to model the conformational landscape of macrocyclic peptides have the potential to enable rational design and optimization. However, accurate, fast, and scalable methods for modeling macrocycle geometries remain elusive. Recent deep learning approaches have significantly accelerated protein structure prediction and the generation of small-molecule conformational ensembles, yet similar progress has not been made for macrocyclic peptides due to their unique properties. Here, we introduce CREMP, a resource generated for the rapid development and evaluation of machine learning models for macrocyclic peptides. CREMP contains 36,198 unique macrocyclic peptides and their high-quality structural ensembles generated using the Conformer-Rotamer Ensemble Sampling Tool (CREST). Altogether, this new dataset contains nearly 31.3 million unique macrocycle geometries, each annotated with energies derived from semi-empirical extended tight-binding (xTB) DFT calculations. We anticipate that this dataset will enable the development of machine learning models that can improve peptide design and optimization for novel therapeutics.
Improving Graph Generation by Restricting Graph Bandwidth
Jan 25, 2023



Deep graph generative modeling has proven capable of learning the distribution of complex, multi-scale structures characterizing real-world graphs. However, one of the main limitations of existing methods is their large output space, which limits generation scalability and hinders accurate modeling of the underlying distribution. To overcome these limitations, we propose a novel approach that significantly reduces the output space of existing graph generative models. Specifically, starting from the observation that many real-world graphs have low graph bandwidth, we restrict graph bandwidth during training and generation. Our strategy improves both generation scalability and quality without increasing architectural complexity or reducing expressiveness. Our approach is compatible with existing graph generative methods, and we describe its application to both autoregressive and one-shot models. We extensively validate our strategy on synthetic and real datasets, including molecular graphs. Our experiments show that, in addition to improving generation efficiency, our approach consistently improves generation quality and reconstruction accuracy. The implementation is made available.
A 3D-Shape Similarity-based Contrastive Approach to Molecular Representation Learning
Nov 03, 2022Molecular shape and geometry dictate key biophysical recognition processes, yet many graph neural networks disregard 3D information for molecular property prediction. Here, we propose a new contrastive-learning procedure for graph neural networks, Molecular Contrastive Learning from Shape Similarity (MolCLaSS), that implicitly learns a three-dimensional representation. Rather than directly encoding or targeting three-dimensional poses, MolCLaSS matches a similarity objective based on Gaussian overlays to learn a meaningful representation of molecular shape. We demonstrate how this framework naturally captures key aspects of three-dimensionality that two-dimensional representations cannot and provides an inductive framework for scaffold hopping.
Attention-Based Learning on Molecular Ensembles
Nov 25, 2020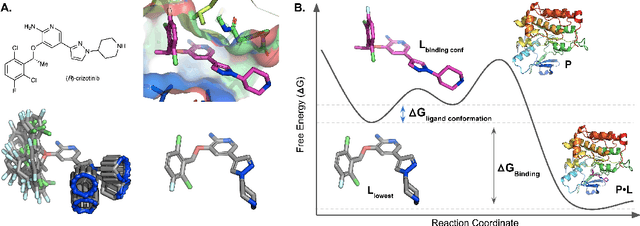
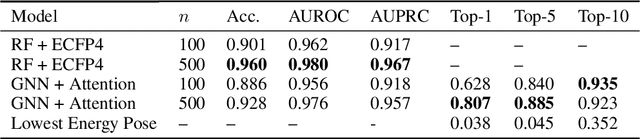
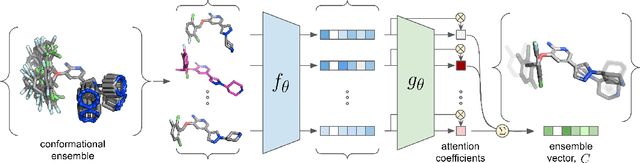
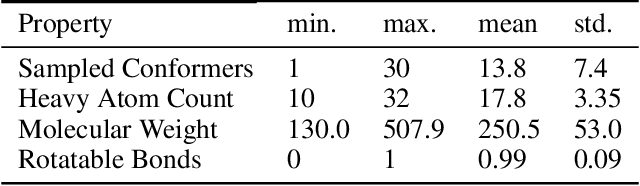
The three-dimensional shape and conformation of small-molecule ligands are critical for biomolecular recognition, yet encoding 3D geometry has not improved ligand-based virtual screening approaches. We describe an end-to-end deep learning approach that operates directly on small-molecule conformational ensembles and identifies key conformational poses of small-molecules. Our networks leverage two levels of representation learning: 1) individual conformers are first encoded as spatial graphs using a graph neural network, and 2) sampled conformational ensembles are represented as sets using an attention mechanism to aggregate over individual instances. We demonstrate the feasibility of this approach on a simple task based on bidentate coordination of biaryl ligands, and show how attention-based pooling can elucidate key conformational poses in tasks based on molecular geometry. This work illustrates how set-based learning approaches may be further developed for small molecule-based virtual screening.