 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgescCBGM: Interpretable Single-Cell Counterfactual Editing
Jun 05, 2026Understanding cellular phenotypes and how they respond to perturbations is critical for disease biology and therapeutic design. Single-cell RNA sequencing enables characterization at cellular resolution, yet the combinatorial space of conditions makes exhaustive experimental mapping infeasible. We introduce single-cell Concept Bottleneck Generative Models (scCBGM), a framework for interpretable and precise counterfactual editing of individual cells. scCBGM adapts concept bottleneck architectures for single-cell data through decoder skip connections and a cross-covariance penalty that promotes disentanglement without dimensional constraints. We extend the framework to flow matching models, enabling concept-guided editing in both encoding-decoding and generation regimes. To enable rigorous evaluation, we develop a synthetic benchmark with ground-truth counterfactuals. Across multiple real datasets, scCBGM demonstrates superior performance in combinatorial generalization and counterfactual prediction, supported by cell-level validation on synthetic data and population-level benchmarks on real datasets.
Generate in Reconstruction Space, Match in Semantic Space: Transport Geometry for One-Step Generation
May 30, 2026Generative modeling and self-supervised representation learning (SSL) optimize structurally different objectives: generative training rewards distributional fidelity, while SSL rewards semantic coherence. Yet recent work repeatedly finds that SSL features improve generative training, though the mechanism of this synergy remains unclear. Here, we study the benefits of SSL in generative modeling in the framework of one-step generation where the role of representation is explicit: frozen SSL features are used to match generated samples to real data. We use the Sinkhorn divergence in that feature space, providing a tractable surrogate for the Wasserstein distance, the population-level discrepancy approximated by Fréchet-style evaluation metrics (such as FID). We find that this objective becomes highly effective when computed in a semantically structured SSL feature space (a 39$\times$ reduction in ImageNet FID). We trace this behavior primarily to matching estimation: semantic SSL features that suppress nuisance reconstruction details induce a more compact geometry, making distribution matching more tractable. As a consequence, the best training SSL features need not match the features used by the evaluation metric. In particular, we show that using Inception as the feature extractor can improve FID while degrading matching stability and sample quality, revealing a form of metric hacking. Using extensive experiments on ImageNet, we identify which SSL feature families lead to best generation performance and show that matching stability is a quantitative criterion for selecting them. Code is available at https://github.com/Genentech/semantic-transport-generation.
AssayBench: An Assay-Level Virtual Cell Benchmark for LLMs and Agents
May 11, 2026Recent advances in machine learning and large-scale biological data collections have revived the prospect of building a virtual cell, a computational model of cellular behavior that could accelerate biological discovery. One of the most compelling promises of this vision is the ability to perform in silico phenotypic screens, in which a model predicts the effects of cellular perturbations in unseen biological contexts. This task combines heterogeneous textual inputs with diverse phenotypic outputs, making it particularly well-suited to LLMs and agentic systems. Yet, no standard benchmark currently exists for this task, as existing efforts focus on narrower molecular readouts that are only indirectly aligned with the phenotypic endpoints driving many real-world drug discovery workflows. In this work, we present AssayBench, a benchmark for phenotypic screen prediction, built from 1,920 publicly available CRISPR screens spanning five broad classes of cellular phenotypes. We formulate the screen prediction task as a gene rank prediction for each screen and introduce the adjusted nDCG, a continuous metric for comparing performance across heterogeneous assays. Our extensive evaluation shows that existing methods remain far from empirically estimated performance ceilings and zero-shot generalist LLMs outperform biology-specific LLMs and trainable baselines. Optimization techniques such as fine-tuning, ensembling, and prompt optimization can further improve LLM performance on this task. Overall, AssayBench offers a practical testbed for measuring progress toward in silico phenotypic screening and, more broadly, virtual cell models.
DC-W2S: Dual-Consensus Weak-to-Strong Training for Reliable Process Reward Modeling in Biological Reasoning
Mar 09, 2026In scientific reasoning tasks, the veracity of the reasoning process is as critical as the final outcome. While Process Reward Models (PRMs) offer a solution to the coarse-grained supervision problems inherent in Outcome Reward Models (ORMs), their deployment is hindered by the prohibitive cost of obtaining expert-verified step-wise labels. This paper addresses the challenge of training reliable PRMs using abundant but noisy "weak" supervision. We argue that existing Weak-to-Strong Generalization (W2SG) theories lack prescriptive guidelines for selecting high-quality training signals from noisy data. To bridge this gap, we introduce the Dual-Consensus Weak-to-Strong (DC-W2S) framework. By intersecting Self-Consensus (SC) metrics among weak supervisors with Neighborhood-Consensus (NC) metrics in the embedding space, we stratify supervision signals into distinct reliability regimes. We then employ a curriculum of instance-level balanced sampling and label-level reliability-aware masking to guide the training process. We demonstrate that DC-W2S enables the training of robust PRMs for complex reasoning without exhaustive expert annotation, proving that strategic data curation is more effective than indiscriminate training on large-scale noisy datasets.
Dynamic Search for Inference-Time Alignment in Diffusion Models
Mar 03, 2025
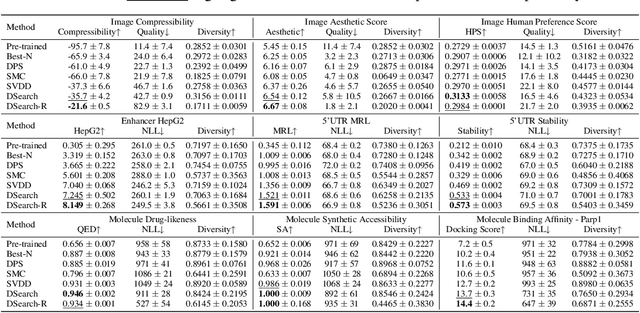
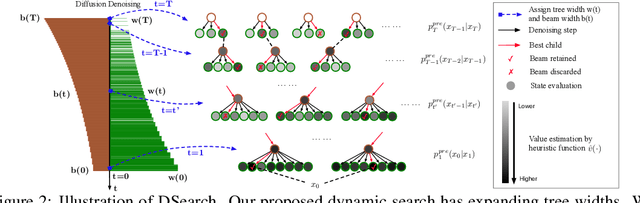

Diffusion models have shown promising generative capabilities across diverse domains, yet aligning their outputs with desired reward functions remains a challenge, particularly in cases where reward functions are non-differentiable. Some gradient-free guidance methods have been developed, but they often struggle to achieve optimal inference-time alignment. In this work, we newly frame inference-time alignment in diffusion as a search problem and propose Dynamic Search for Diffusion (DSearch), which subsamples from denoising processes and approximates intermediate node rewards. It also dynamically adjusts beam width and tree expansion to efficiently explore high-reward generations. To refine intermediate decisions, DSearch incorporates adaptive scheduling based on noise levels and a lookahead heuristic function. We validate DSearch across multiple domains, including biological sequence design, molecular optimization, and image generation, demonstrating superior reward optimization compared to existing approaches.
RAG-Enhanced Collaborative LLM Agents for Drug Discovery
Feb 22, 2025Recent advances in large language models (LLMs) have shown great potential to accelerate drug discovery. However, the specialized nature of biochemical data often necessitates costly domain-specific fine-tuning, posing critical challenges. First, it hinders the application of more flexible general-purpose LLMs in cutting-edge drug discovery tasks. More importantly, it impedes the rapid integration of the vast amounts of scientific data continuously generated through experiments and research. To investigate these challenges, we propose CLADD, a retrieval-augmented generation (RAG)-empowered agentic system tailored to drug discovery tasks. Through the collaboration of multiple LLM agents, CLADD dynamically retrieves information from biomedical knowledge bases, contextualizes query molecules, and integrates relevant evidence to generate responses -- all without the need for domain-specific fine-tuning. Crucially, we tackle key obstacles in applying RAG workflows to biochemical data, including data heterogeneity, ambiguity, and multi-source integration. We demonstrate the flexibility and effectiveness of this framework across a variety of drug discovery tasks, showing that it outperforms general-purpose and domain-specific LLMs as well as traditional deep learning approaches.
Efficient Fine-Tuning of Single-Cell Foundation Models Enables Zero-Shot Molecular Perturbation Prediction
Dec 18, 2024



Predicting transcriptional responses to novel drugs provides a unique opportunity to accelerate biomedical research and advance drug discovery efforts. However, the inherent complexity and high dimensionality of cellular responses, combined with the extremely limited available experimental data, makes the task challenging. In this study, we leverage single-cell foundation models (FMs) pre-trained on tens of millions of single cells, encompassing multiple cell types, states, and disease annotations, to address molecular perturbation prediction. We introduce a drug-conditional adapter that allows efficient fine-tuning by training less than 1% of the original foundation model, thus enabling molecular conditioning while preserving the rich biological representation learned during pre-training. The proposed strategy allows not only the prediction of cellular responses to novel drugs, but also the zero-shot generalization to unseen cell lines. We establish a robust evaluation framework to assess model performance across different generalization tasks, demonstrating state-of-the-art results across all settings, with significant improvements in the few-shot and zero-shot generalization to new cell lines compared to existing baselines.
MolCap-Arena: A Comprehensive Captioning Benchmark on Language-Enhanced Molecular Property Prediction
Nov 01, 2024



Bridging biomolecular modeling with natural language information, particularly through large language models (LLMs), has recently emerged as a promising interdisciplinary research area. LLMs, having been trained on large corpora of scientific documents, demonstrate significant potential in understanding and reasoning about biomolecules by providing enriched contextual and domain knowledge. However, the extent to which LLM-driven insights can improve performance on complex predictive tasks (e.g., toxicity) remains unclear. Further, the extent to which relevant knowledge can be extracted from LLMs also remains unknown. In this study, we present Molecule Caption Arena: the first comprehensive benchmark of LLM-augmented molecular property prediction. We evaluate over twenty LLMs, including both general-purpose and domain-specific molecule captioners, across diverse prediction tasks. To this goal, we introduce a novel, battle-based rating system. Our findings confirm the ability of LLM-extracted knowledge to enhance state-of-the-art molecular representations, with notable model-, prompt-, and dataset-specific variations. Code, resources, and data are available at github.com/Genentech/molcap-arena.
A mechanistically interpretable neural network for regulatory genomics
Oct 08, 2024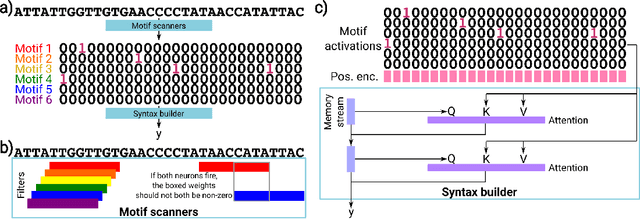
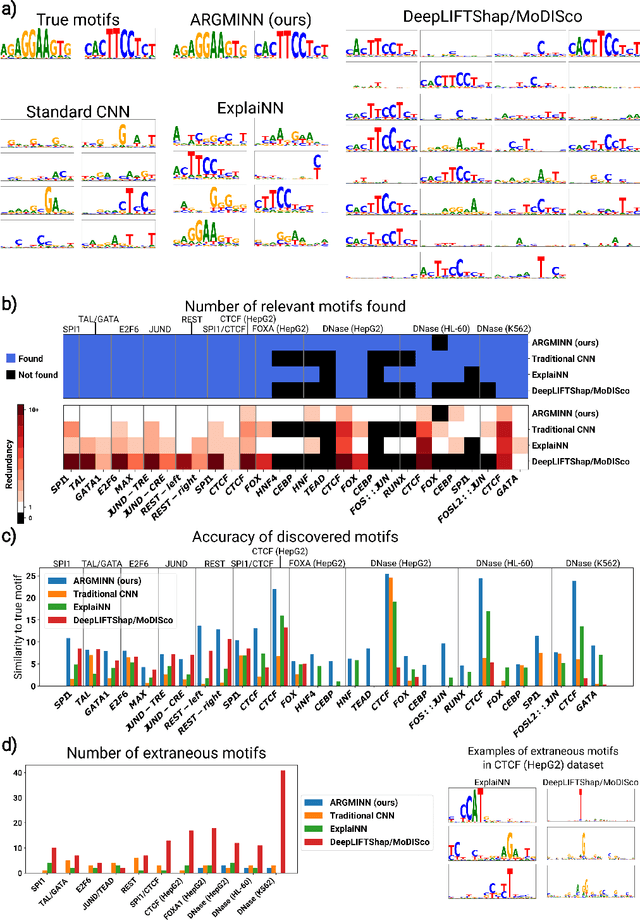

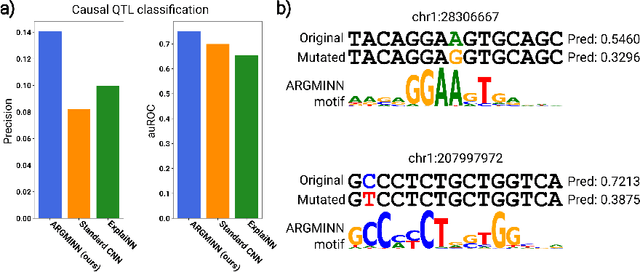
Deep neural networks excel in mapping genomic DNA sequences to associated readouts (e.g., protein-DNA binding). Beyond prediction, the goal of these networks is to reveal to scientists the underlying motifs (and their syntax) which drive genome regulation. Traditional methods that extract motifs from convolutional filters suffer from the uninterpretable dispersion of information across filters and layers. Other methods which rely on importance scores can be unstable and unreliable. Instead, we designed a novel mechanistically interpretable architecture for regulatory genomics, where motifs and their syntax are directly encoded and readable from the learned weights and activations. We provide theoretical and empirical evidence of our architecture's full expressivity, while still being highly interpretable. Through several experiments, we show that our architecture excels in de novo motif discovery and motif instance calling, is robust to variable sequence contexts, and enables fully interpretable generation of novel functional sequences.
Derivative-Free Guidance in Continuous and Discrete Diffusion Models with Soft Value-Based Decoding
Aug 15, 2024



Diffusion models excel at capturing the natural design spaces of images, molecules, DNA, RNA, and protein sequences. However, rather than merely generating designs that are natural, we often aim to optimize downstream reward functions while preserving the naturalness of these design spaces. Existing methods for achieving this goal often require ``differentiable'' proxy models (\textit{e.g.}, classifier guidance or DPS) or involve computationally expensive fine-tuning of diffusion models (\textit{e.g.}, classifier-free guidance, RL-based fine-tuning). In our work, we propose a new method to address these challenges. Our algorithm is an iterative sampling method that integrates soft value functions, which looks ahead to how intermediate noisy states lead to high rewards in the future, into the standard inference procedure of pre-trained diffusion models. Notably, our approach avoids fine-tuning generative models and eliminates the need to construct differentiable models. This enables us to (1) directly utilize non-differentiable features/reward feedback, commonly used in many scientific domains, and (2) apply our method to recent discrete diffusion models in a principled way. Finally, we demonstrate the effectiveness of our algorithm across several domains, including image generation, molecule generation, and DNA/RNA sequence generation. The code is available at \href{https://github.com/masa-ue/SVDD}{https://github.com/masa-ue/SVDD}.