 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGroup Contrastive Learning for Weakly Paired Multimodal Data
Feb 03, 2026We present GROOVE, a semi-supervised multi-modal representation learning approach for high-content perturbation data where samples across modalities are weakly paired through shared perturbation labels but lack direct correspondence. Our primary contribution is GroupCLIP, a novel group-level contrastive loss that bridges the gap between CLIP for paired cross-modal data and SupCon for uni-modal supervised contrastive learning, addressing a fundamental gap in contrastive learning for weakly-paired settings. We integrate GroupCLIP with an on-the-fly backtranslating autoencoder framework to encourage cross-modally entangled representations while maintaining group-level coherence within a shared latent space. Critically, we introduce a comprehensive combinatorial evaluation framework that systematically assesses representation learners across multiple optimal transport aligners, addressing key limitations in existing evaluation strategies. This framework includes novel simulations that systematically vary shared versus modality-specific perturbation effects enabling principled assessment of method robustness. Our combinatorial benchmarking reveals that there is not yet an aligner that uniformly dominates across settings or modality pairs. Across simulations and two real single-cell genetic perturbation datasets, GROOVE performs on par with or outperforms existing approaches for downstream cross-modal matching and imputation tasks. Our ablation studies demonstrate that GroupCLIP is the key component driving performance gains. These results highlight the importance of leveraging group-level constraints for effective multi-modal representation learning in scenarios where only weak pairing is available.
Contextualizing biological perturbation experiments through language
Feb 28, 2025
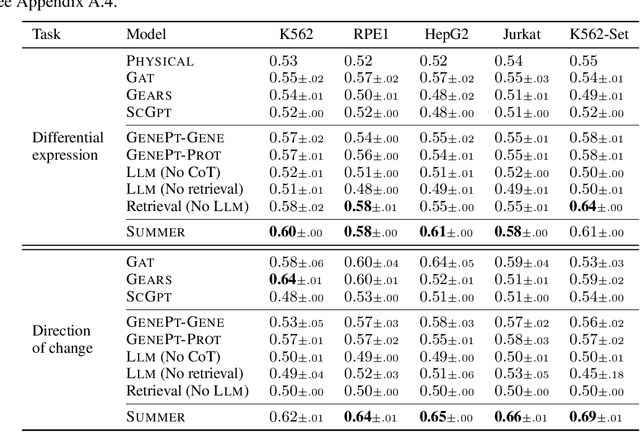

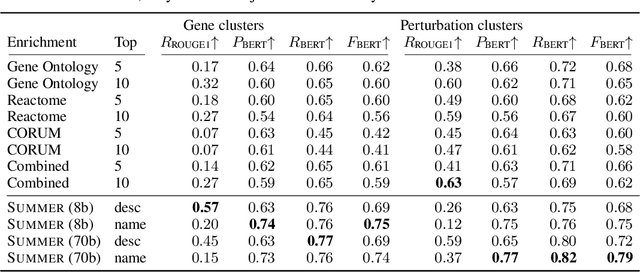
High-content perturbation experiments allow scientists to probe biomolecular systems at unprecedented resolution, but experimental and analysis costs pose significant barriers to widespread adoption. Machine learning has the potential to guide efficient exploration of the perturbation space and extract novel insights from these data. However, current approaches neglect the semantic richness of the relevant biology, and their objectives are misaligned with downstream biological analyses. In this paper, we hypothesize that large language models (LLMs) present a natural medium for representing complex biological relationships and rationalizing experimental outcomes. We propose PerturbQA, a benchmark for structured reasoning over perturbation experiments. Unlike current benchmarks that primarily interrogate existing knowledge, PerturbQA is inspired by open problems in perturbation modeling: prediction of differential expression and change of direction for unseen perturbations, and gene set enrichment. We evaluate state-of-the-art machine learning and statistical approaches for modeling perturbations, as well as standard LLM reasoning strategies, and we find that current methods perform poorly on PerturbQA. As a proof of feasibility, we introduce Summer (SUMMarize, retrievE, and answeR, a simple, domain-informed LLM framework that matches or exceeds the current state-of-the-art. Our code and data are publicly available at https://github.com/genentech/PerturbQA.
Supervised Contrastive Block Disentanglement
Feb 11, 2025



Real-world datasets often combine data collected under different experimental conditions. This yields larger datasets, but also introduces spurious correlations that make it difficult to model the phenomena of interest. We address this by learning two embeddings to independently represent the phenomena of interest and the spurious correlations. The embedding representing the phenomena of interest is correlated with the target variable $y$, and is invariant to the environment variable $e$. In contrast, the embedding representing the spurious correlations is correlated with $e$. The invariance to $e$ is difficult to achieve on real-world datasets. Our primary contribution is an algorithm called Supervised Contrastive Block Disentanglement (SCBD) that effectively enforces this invariance. It is based purely on Supervised Contrastive Learning, and applies to real-world data better than existing approaches. We empirically validate SCBD on two challenging problems. The first problem is domain generalization, where we achieve strong performance on a synthetic dataset, as well as on Camelyon17-WILDS. We introduce a single hyperparameter $\alpha$ to control the degree of invariance to $e$. When we increase $\alpha$ to strengthen the degree of invariance, out-of-distribution performance improves at the expense of in-distribution performance. The second problem is batch correction, in which we apply SCBD to preserve biological signal and remove inter-well batch effects when modeling single-cell perturbations from 26 million Optical Pooled Screening images.
Efficient Fine-Tuning of Single-Cell Foundation Models Enables Zero-Shot Molecular Perturbation Prediction
Dec 18, 2024



Predicting transcriptional responses to novel drugs provides a unique opportunity to accelerate biomedical research and advance drug discovery efforts. However, the inherent complexity and high dimensionality of cellular responses, combined with the extremely limited available experimental data, makes the task challenging. In this study, we leverage single-cell foundation models (FMs) pre-trained on tens of millions of single cells, encompassing multiple cell types, states, and disease annotations, to address molecular perturbation prediction. We introduce a drug-conditional adapter that allows efficient fine-tuning by training less than 1% of the original foundation model, thus enabling molecular conditioning while preserving the rich biological representation learned during pre-training. The proposed strategy allows not only the prediction of cellular responses to novel drugs, but also the zero-shot generalization to unseen cell lines. We establish a robust evaluation framework to assess model performance across different generalization tasks, demonstrating state-of-the-art results across all settings, with significant improvements in the few-shot and zero-shot generalization to new cell lines compared to existing baselines.
Learning Identifiable Factorized Causal Representations of Cellular Responses
Oct 29, 2024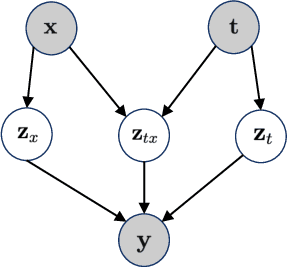
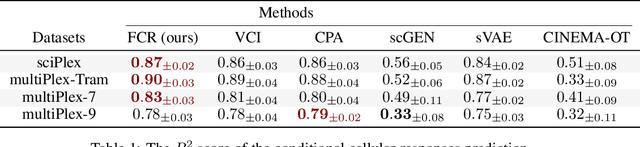
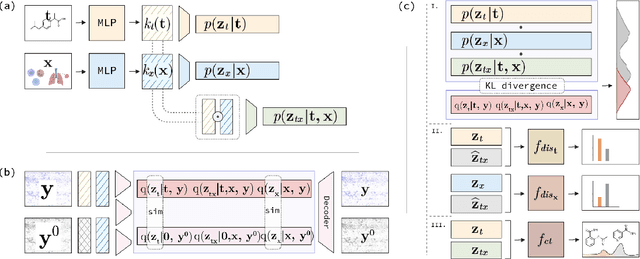

The study of cells and their responses to genetic or chemical perturbations promises to accelerate the discovery of therapeutic targets. However, designing adequate and insightful models for such data is difficult because the response of a cell to perturbations essentially depends on its biological context (e.g., genetic background or cell type). For example, while discovering therapeutic targets, one may want to enrich for drugs that specifically target a certain cell type. This challenge emphasizes the need for methods that explicitly take into account potential interactions between drugs and contexts. Towards this goal, we propose a novel Factorized Causal Representation (FCR) learning method that reveals causal structure in single-cell perturbation data from several cell lines. Based on the framework of identifiable deep generative models, FCR learns multiple cellular representations that are disentangled, comprised of covariate-specific ($\mathbf{z}_x$), treatment-specific ($\mathbf{z}_{t}$), and interaction-specific ($\mathbf{z}_{tx}$) blocks. Based on recent advances in non-linear ICA theory, we prove the component-wise identifiability of $\mathbf{z}_{tx}$ and block-wise identifiability of $\mathbf{z}_t$ and $\mathbf{z}_x$. Then, we present our implementation of FCR, and empirically demonstrate that it outperforms state-of-the-art baselines in various tasks across four single-cell datasets.
Weakly Supervised Set-Consistency Learning Improves Morphological Profiling of Single-Cell Images
Jun 08, 2024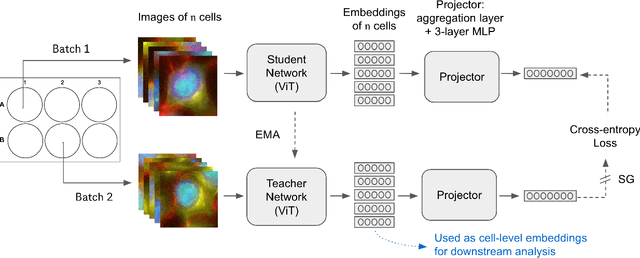



Optical Pooled Screening (OPS) is a powerful tool combining high-content microscopy with genetic engineering to investigate gene function in disease. The characterization of high-content images remains an active area of research and is currently undergoing rapid innovation through the application of self-supervised learning and vision transformers. In this study, we propose a set-level consistency learning algorithm, Set-DINO, that combines self-supervised learning with weak supervision to improve learned representations of perturbation effects in single-cell images. Our method leverages the replicate structure of OPS experiments (i.e., cells undergoing the same genetic perturbation, both within and across batches) as a form of weak supervision. We conduct extensive experiments on a large-scale OPS dataset with more than 5000 genetic perturbations, and demonstrate that Set-DINO helps mitigate the impact of confounders and encodes more biologically meaningful information. In particular, Set-DINO recalls known biological relationships with higher accuracy compared to commonly used methods for morphological profiling, suggesting that it can generate more reliable insights from drug target discovery campaigns leveraging OPS.
Toward the Identifiability of Comparative Deep Generative Models
Jan 29, 2024



Deep Generative Models (DGMs) are versatile tools for learning data representations while adequately incorporating domain knowledge such as the specification of conditional probability distributions. Recently proposed DGMs tackle the important task of comparing data sets from different sources. One such example is the setting of contrastive analysis that focuses on describing patterns that are enriched in a target data set compared to a background data set. The practical deployment of those models often assumes that DGMs naturally infer interpretable and modular latent representations, which is known to be an issue in practice. Consequently, existing methods often rely on ad-hoc regularization schemes, although without any theoretical grounding. Here, we propose a theory of identifiability for comparative DGMs by extending recent advances in the field of non-linear independent component analysis. We show that, while these models lack identifiability across a general class of mixing functions, they surprisingly become identifiable when the mixing function is piece-wise affine (e.g., parameterized by a ReLU neural network). We also investigate the impact of model misspecification, and empirically show that previously proposed regularization techniques for fitting comparative DGMs help with identifiability when the number of latent variables is not known in advance. Finally, we introduce a novel methodology for fitting comparative DGMs that improves the treatment of multiple data sources via multi-objective optimization and that helps adjust the hyperparameter for the regularization in an interpretable manner, using constrained optimization. We empirically validate our theory and new methodology using simulated data as well as a recent data set of genetic perturbations in cells profiled via single-cell RNA sequencing.
* 45 pages, 3 figures