 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSupervised Contrastive Block Disentanglement
Feb 11, 2025



Real-world datasets often combine data collected under different experimental conditions. This yields larger datasets, but also introduces spurious correlations that make it difficult to model the phenomena of interest. We address this by learning two embeddings to independently represent the phenomena of interest and the spurious correlations. The embedding representing the phenomena of interest is correlated with the target variable $y$, and is invariant to the environment variable $e$. In contrast, the embedding representing the spurious correlations is correlated with $e$. The invariance to $e$ is difficult to achieve on real-world datasets. Our primary contribution is an algorithm called Supervised Contrastive Block Disentanglement (SCBD) that effectively enforces this invariance. It is based purely on Supervised Contrastive Learning, and applies to real-world data better than existing approaches. We empirically validate SCBD on two challenging problems. The first problem is domain generalization, where we achieve strong performance on a synthetic dataset, as well as on Camelyon17-WILDS. We introduce a single hyperparameter $\alpha$ to control the degree of invariance to $e$. When we increase $\alpha$ to strengthen the degree of invariance, out-of-distribution performance improves at the expense of in-distribution performance. The second problem is batch correction, in which we apply SCBD to preserve biological signal and remove inter-well batch effects when modeling single-cell perturbations from 26 million Optical Pooled Screening images.
Learning Identifiable Factorized Causal Representations of Cellular Responses
Oct 29, 2024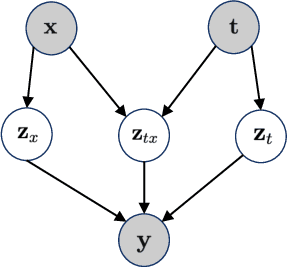
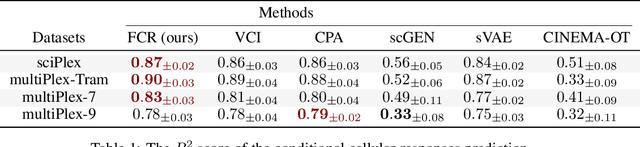
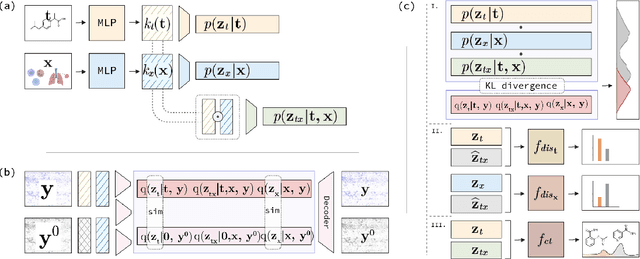

The study of cells and their responses to genetic or chemical perturbations promises to accelerate the discovery of therapeutic targets. However, designing adequate and insightful models for such data is difficult because the response of a cell to perturbations essentially depends on its biological context (e.g., genetic background or cell type). For example, while discovering therapeutic targets, one may want to enrich for drugs that specifically target a certain cell type. This challenge emphasizes the need for methods that explicitly take into account potential interactions between drugs and contexts. Towards this goal, we propose a novel Factorized Causal Representation (FCR) learning method that reveals causal structure in single-cell perturbation data from several cell lines. Based on the framework of identifiable deep generative models, FCR learns multiple cellular representations that are disentangled, comprised of covariate-specific ($\mathbf{z}_x$), treatment-specific ($\mathbf{z}_{t}$), and interaction-specific ($\mathbf{z}_{tx}$) blocks. Based on recent advances in non-linear ICA theory, we prove the component-wise identifiability of $\mathbf{z}_{tx}$ and block-wise identifiability of $\mathbf{z}_t$ and $\mathbf{z}_x$. Then, we present our implementation of FCR, and empirically demonstrate that it outperforms state-of-the-art baselines in various tasks across four single-cell datasets.
Gene-Level Representation Learning via Interventional Style Transfer in Optical Pooled Screening
Jun 11, 2024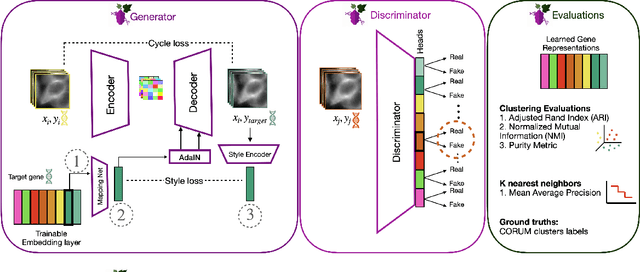
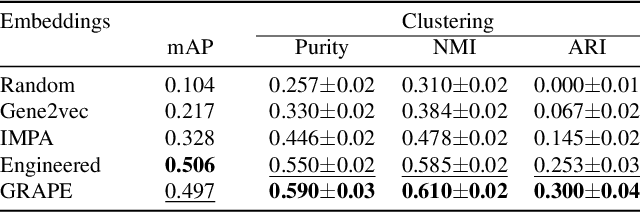
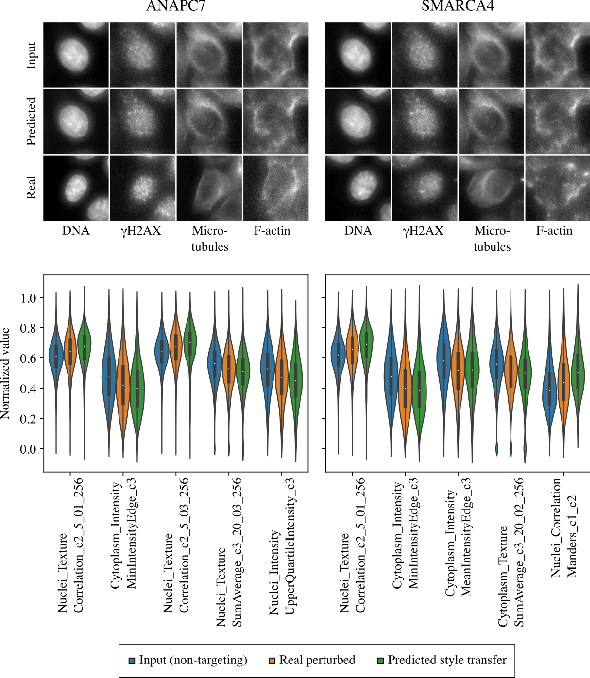

Optical pooled screening (OPS) combines automated microscopy and genetic perturbations to systematically study gene function in a scalable and cost-effective way. Leveraging the resulting data requires extracting biologically informative representations of cellular perturbation phenotypes from images. We employ a style-transfer approach to learn gene-level feature representations from images of genetically perturbed cells obtained via OPS. Our method outperforms widely used engineered features in clustering gene representations according to gene function, demonstrating its utility for uncovering latent biological relationships. This approach offers a promising alternative to investigate the role of genes in health and disease.
Toward the Identifiability of Comparative Deep Generative Models
Jan 29, 2024



Deep Generative Models (DGMs) are versatile tools for learning data representations while adequately incorporating domain knowledge such as the specification of conditional probability distributions. Recently proposed DGMs tackle the important task of comparing data sets from different sources. One such example is the setting of contrastive analysis that focuses on describing patterns that are enriched in a target data set compared to a background data set. The practical deployment of those models often assumes that DGMs naturally infer interpretable and modular latent representations, which is known to be an issue in practice. Consequently, existing methods often rely on ad-hoc regularization schemes, although without any theoretical grounding. Here, we propose a theory of identifiability for comparative DGMs by extending recent advances in the field of non-linear independent component analysis. We show that, while these models lack identifiability across a general class of mixing functions, they surprisingly become identifiable when the mixing function is piece-wise affine (e.g., parameterized by a ReLU neural network). We also investigate the impact of model misspecification, and empirically show that previously proposed regularization techniques for fitting comparative DGMs help with identifiability when the number of latent variables is not known in advance. Finally, we introduce a novel methodology for fitting comparative DGMs that improves the treatment of multiple data sources via multi-objective optimization and that helps adjust the hyperparameter for the regularization in an interpretable manner, using constrained optimization. We empirically validate our theory and new methodology using simulated data as well as a recent data set of genetic perturbations in cells profiled via single-cell RNA sequencing.
* 45 pages, 3 figures
NODAGS-Flow: Nonlinear Cyclic Causal Structure Learning
Jan 04, 2023Learning causal relationships between variables is a well-studied problem in statistics, with many important applications in science. However, modeling real-world systems remain challenging, as most existing algorithms assume that the underlying causal graph is acyclic. While this is a convenient framework for developing theoretical developments about causal reasoning and inference, the underlying modeling assumption is likely to be violated in real systems, because feedback loops are common (e.g., in biological systems). Although a few methods search for cyclic causal models, they usually rely on some form of linearity, which is also limiting, or lack a clear underlying probabilistic model. In this work, we propose a novel framework for learning nonlinear cyclic causal graphical models from interventional data, called NODAGS-Flow. We perform inference via direct likelihood optimization, employing techniques from residual normalizing flows for likelihood estimation. Through synthetic experiments and an application to single-cell high-content perturbation screening data, we show significant performance improvements with our approach compared to state-of-the-art methods with respect to structure recovery and predictive performance.
Learning Causal Representations of Single Cells via Sparse Mechanism Shift Modeling
Nov 09, 2022



Latent variable models such as the Variational Auto-Encoder (VAE) have become a go-to tool for analyzing biological data, especially in the field of single-cell genomics. One remaining challenge is the interpretability of latent variables as biological processes that define a cell's identity. Outside of biological applications, this problem is commonly referred to as learning disentangled representations. Although several disentanglement-promoting variants of the VAE were introduced, and applied to single-cell genomics data, this task has been shown to be infeasible from independent and identically distributed measurements, without additional structure. Instead, recent methods propose to leverage non-stationary data, as well as the sparse mechanism shift assumption in order to learn disentangled representations with a causal semantic. Here, we extend the application of these methodological advances to the analysis of single-cell genomics data with genetic or chemical perturbations. More precisely, we propose a deep generative model of single-cell gene expression data for which each perturbation is treated as a stochastic intervention targeting an unknown, but sparse, subset of latent variables. We benchmark these methods on simulated single-cell data to evaluate their performance at latent units recovery, causal target identification and out-of-domain generalization. Finally, we apply those approaches to two real-world large-scale gene perturbation data sets and find that models that exploit the sparse mechanism shift hypothesis surpass contemporary methods on a transfer learning task. We implement our new model and benchmarks using the scvi-tools library, and release it as open-source software at https://github.com/Genentech/sVAE.
Large-Scale Differentiable Causal Discovery of Factor Graphs
Jun 15, 2022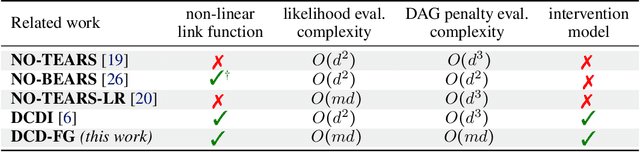
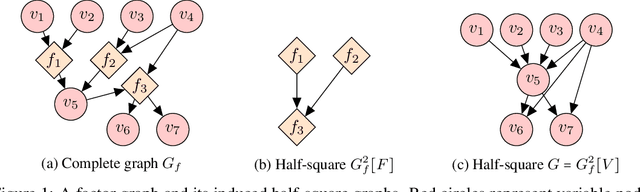
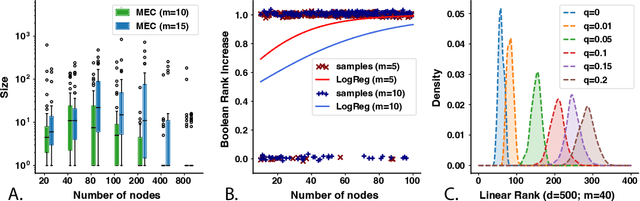

A common theme in causal inference is learning causal relationships between observed variables, also known as causal discovery. This is usually a daunting task, given the large number of candidate causal graphs and the combinatorial nature of the search space. Perhaps for this reason, most research has so far focused on relatively small causal graphs, with up to hundreds of nodes. However, recent advances in fields like biology enable generating experimental data sets with thousands of interventions followed by rich profiling of thousands of variables, raising the opportunity and urgent need for large causal graph models. Here, we introduce the notion of factor directed acyclic graphs (f-DAGs) as a way to restrict the search space to non-linear low-rank causal interaction models. Combining this novel structural assumption with recent advances that bridge the gap between causal discovery and continuous optimization, we achieve causal discovery on thousands of variables. Additionally, as a model for the impact of statistical noise on this estimation procedure, we study a model of edge perturbations of the f-DAG skeleton based on random graphs and quantify the effect of such perturbations on the f-DAG rank. This theoretical analysis suggests that the set of candidate f-DAGs is much smaller than the whole DAG space and thus more statistically robust in the high-dimensional regime where the underlying skeleton is hard to assess. We propose Differentiable Causal Discovery of Factor Graphs (DCD-FG), a scalable implementation of f-DAG constrained causal discovery for high-dimensional interventional data. DCD-FG uses a Gaussian non-linear low-rank structural equation model and shows significant improvements compared to state-of-the-art methods in both simulations as well as a recent large-scale single-cell RNA sequencing data set with hundreds of genetic interventions.
Learning from eXtreme Bandit Feedback
Sep 27, 2020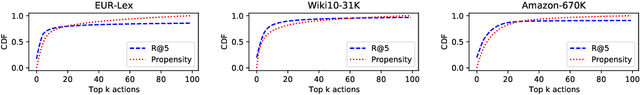
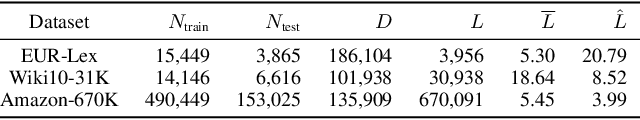
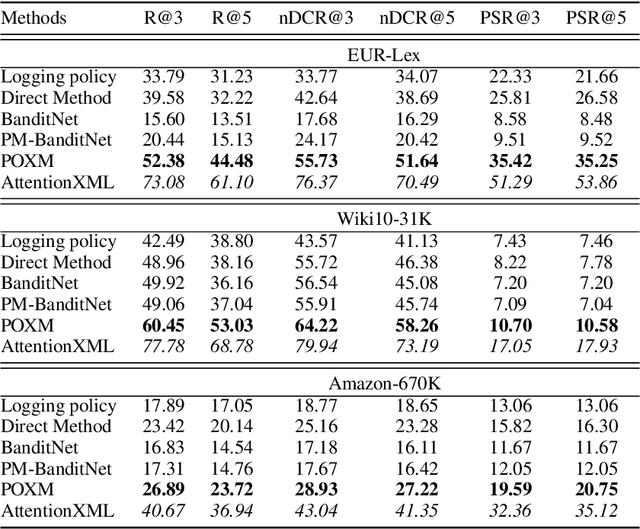
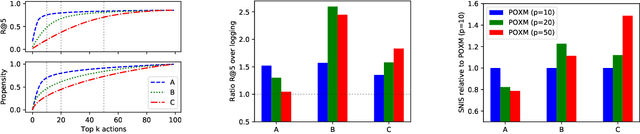
We study the problem of batch learning from bandit feedback in the setting of extremely large action spaces. Learning from extreme bandit feedback is ubiquitous in recommendation systems, in which billions of decisions are made over sets consisting of millions of choices in a single day, yielding massive observational data. In these large-scale real-world applications, supervised learning frameworks such as eXtreme Multi-label Classification (XMC) are widely used despite the fact that they incur significant biases due to the mismatch between bandit feedback and supervised labels. Such biases can be mitigated by importance sampling techniques, but these techniques suffer from impractical variance when dealing with a large number of actions. In this paper, we introduce a selective importance sampling estimator (sIS) that operates in a significantly more favorable bias-variance regime. The sIS estimator is obtained by performing importance sampling on the conditional expectation of the reward with respect to a small subset of actions for each instance (a form of Rao-Blackwellization). We employ this estimator in a novel algorithmic procedure---named Policy Optimization for eXtreme Models (POXM)---for learning from bandit feedback on XMC tasks. In POXM, the selected actions for the sIS estimator are the top-p actions of the logging policy, where p is adjusted from the data and is significantly smaller than the size of the action space. We use a supervised-to-bandit conversion on three XMC datasets to benchmark our POXM method against three competing methods: BanditNet, a previously applied partial matching pruning strategy, and a supervised learning baseline. Whereas BanditNet sometimes improves marginally over the logging policy, our experiments show that POXM systematically and significantly improves over all baselines.
Decision-Making with Auto-Encoding Variational Bayes
Feb 17, 2020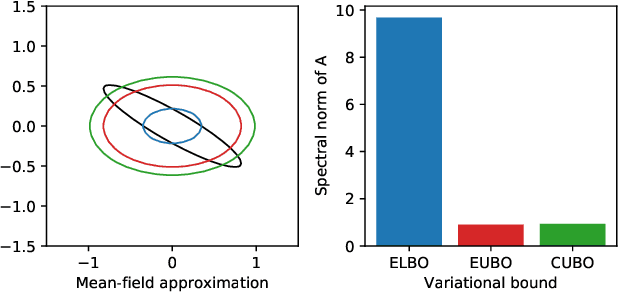

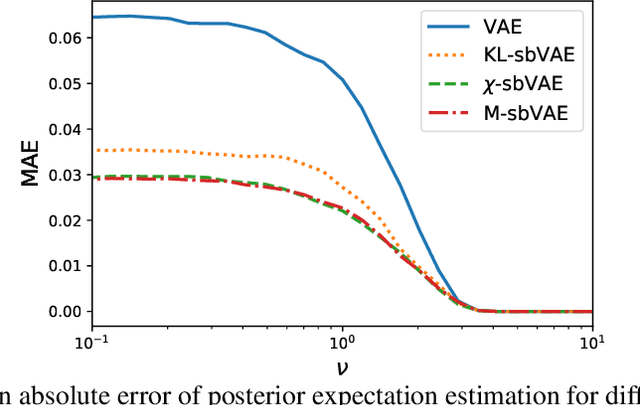

To make decisions based on a model fit by Auto-Encoding Variational Bayes (AEVB), practitioners typically use importance sampling to estimate a functional of the posterior distribution. The variational distribution found by AEVB serves as the proposal distribution for importance sampling. However, this proposal distribution may give unreliable (high variance) importance sampling estimates, thus leading to poor decisions. We explore how changing the objective function for learning the variational distribution, while continuing to learn the generative model based on the ELBO, affects the quality of downstream decisions. For a particular model, we characterize the error of importance sampling as a function of posterior variance and show that proposal distributions learned with evidence upper bounds are better. Motivated by these theoretical results, we propose a novel variant of the VAE. In addition to experimenting with MNIST, we present a full-fledged application of the proposed method to single-cell RNA sequencing. In this challenging instance of multiple hypothesis testing, the proposed method surpasses the current state of the art.
A joint model of unpaired data from scRNA-seq and spatial transcriptomics for imputing missing gene expression measurements
May 06, 2019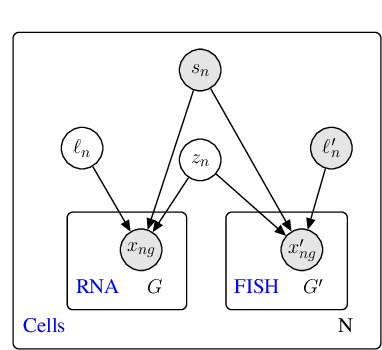
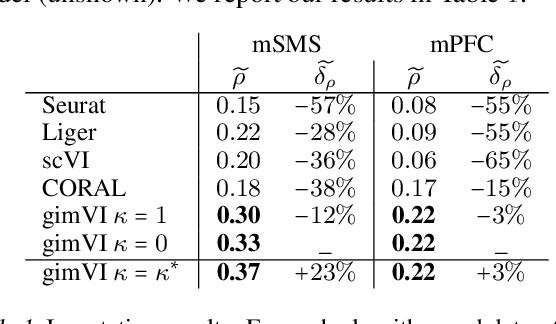
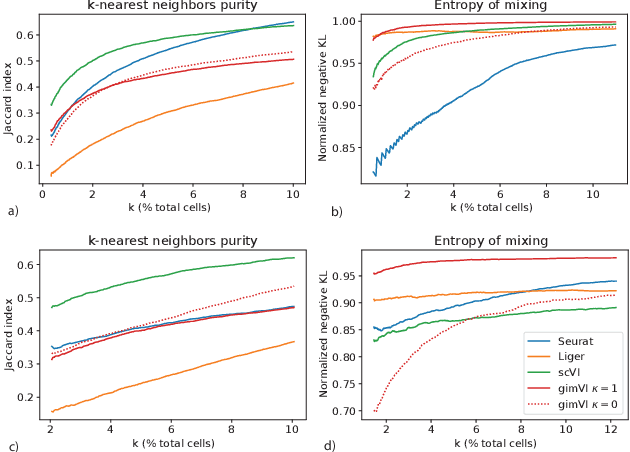
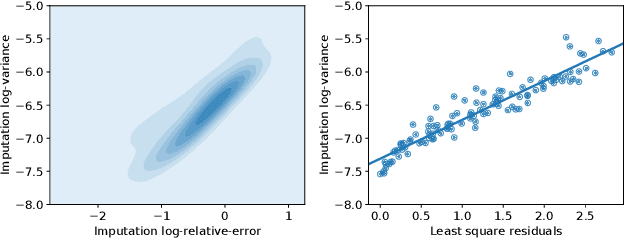
Spatial studies of transcriptome provide biologists with gene expression maps of heterogeneous and complex tissues. However, most experimental protocols for spatial transcriptomics suffer from the need to select beforehand a small fraction of genes to be quantified over the entire transcriptome. Standard single-cell RNA sequencing (scRNA-seq) is more prevalent, easier to implement and can in principle capture any gene but cannot recover the spatial location of the cells. In this manuscript, we focus on the problem of imputation of missing genes in spatial transcriptomic data based on (unpaired) standard scRNA-seq data from the same biological tissue. Building upon domain adaptation work, we propose gimVI, a deep generative model for the integration of spatial transcriptomic data and scRNA-seq data that can be used to impute missing genes. After describing our generative model and an inference procedure for it, we compare gimVI to alternative methods from computational biology or domain adaptation on real datasets and outperform Seurat Anchors, Liger and CORAL to impute held-out genes.