 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLearning Identifiable Factorized Causal Representations of Cellular Responses
Paper and Code
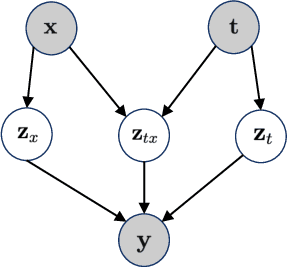
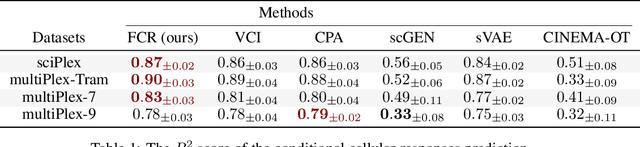
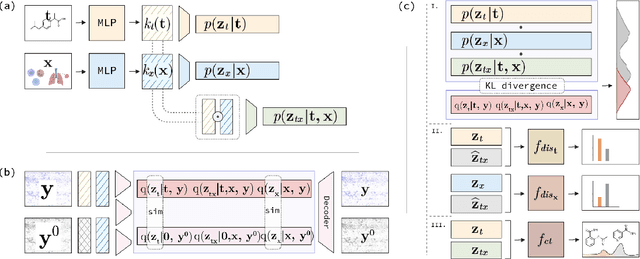

The study of cells and their responses to genetic or chemical perturbations promises to accelerate the discovery of therapeutic targets. However, designing adequate and insightful models for such data is difficult because the response of a cell to perturbations essentially depends on its biological context (e.g., genetic background or cell type). For example, while discovering therapeutic targets, one may want to enrich for drugs that specifically target a certain cell type. This challenge emphasizes the need for methods that explicitly take into account potential interactions between drugs and contexts. Towards this goal, we propose a novel Factorized Causal Representation (FCR) learning method that reveals causal structure in single-cell perturbation data from several cell lines. Based on the framework of identifiable deep generative models, FCR learns multiple cellular representations that are disentangled, comprised of covariate-specific ($\mathbf{z}_x$), treatment-specific ($\mathbf{z}_{t}$), and interaction-specific ($\mathbf{z}_{tx}$) blocks. Based on recent advances in non-linear ICA theory, we prove the component-wise identifiability of $\mathbf{z}_{tx}$ and block-wise identifiability of $\mathbf{z}_t$ and $\mathbf{z}_x$. Then, we present our implementation of FCR, and empirically demonstrate that it outperforms state-of-the-art baselines in various tasks across four single-cell datasets.