 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeWhen to Align, When to Predict: A Phase Diagram for Multimodal Learning
Jun 09, 2026Cross-modal alignment (CA) and cross-modal prediction (CP) are the dominant paradigms for multimodal representation learning, yet there is no systematic understanding of when each succeeds, when each fails, and when cross-modal training helps at all -- a gap that leaves practitioners, especially in scientific domains like biomedicine or astrophysics, with heterogeneous instruments and multiple levels of organization and measurement, unable to diagnose why standard methods underperform the best single modality. We develop a unified linear framework that addresses both questions. Under a spiked signal-plus-noise model with structured cross-modal nuisance correlation, we derive separation ratios for both objectives that expose complementary failure modes: alignment whitens each modality and fails when nuisance is strongly correlated across views; prediction encodes whatever is cross-predictable through a one-sided whitening, with recovery governed by source-modality quality. The resulting phase diagram partitions multimodal problems into four regimes: Both, CA only, CP only, and Neither. We present a data-driven procedure to locate real-world datasets in this diagram using a small labeled subsample, identifying the preferred objective and prediction direction before any cross-modal training. Experiments on synthetic data, stereo-vision benchmarks, image-caption pairs, and real astrophysical data validate the predictions in the nonlinear regime, including the Neither regime where cross-modal training is actively harmful. Our framework lets practitioners diagnose their multimodal problem and choose the right objective before committing to training. Code to reproduce the results is available at https://github.com/IlayMalinyak/mm_align_vs_pred.
Generate in Reconstruction Space, Match in Semantic Space: Transport Geometry for One-Step Generation
May 30, 2026Generative modeling and self-supervised representation learning (SSL) optimize structurally different objectives: generative training rewards distributional fidelity, while SSL rewards semantic coherence. Yet recent work repeatedly finds that SSL features improve generative training, though the mechanism of this synergy remains unclear. Here, we study the benefits of SSL in generative modeling in the framework of one-step generation where the role of representation is explicit: frozen SSL features are used to match generated samples to real data. We use the Sinkhorn divergence in that feature space, providing a tractable surrogate for the Wasserstein distance, the population-level discrepancy approximated by Fréchet-style evaluation metrics (such as FID). We find that this objective becomes highly effective when computed in a semantically structured SSL feature space (a 39$\times$ reduction in ImageNet FID). We trace this behavior primarily to matching estimation: semantic SSL features that suppress nuisance reconstruction details induce a more compact geometry, making distribution matching more tractable. As a consequence, the best training SSL features need not match the features used by the evaluation metric. In particular, we show that using Inception as the feature extractor can improve FID while degrading matching stability and sample quality, revealing a form of metric hacking. Using extensive experiments on ImageNet, we identify which SSL feature families lead to best generation performance and show that matching stability is a quantitative criterion for selecting them. Code is available at https://github.com/Genentech/semantic-transport-generation.
Group Contrastive Learning for Weakly Paired Multimodal Data
Feb 03, 2026We present GROOVE, a semi-supervised multi-modal representation learning approach for high-content perturbation data where samples across modalities are weakly paired through shared perturbation labels but lack direct correspondence. Our primary contribution is GroupCLIP, a novel group-level contrastive loss that bridges the gap between CLIP for paired cross-modal data and SupCon for uni-modal supervised contrastive learning, addressing a fundamental gap in contrastive learning for weakly-paired settings. We integrate GroupCLIP with an on-the-fly backtranslating autoencoder framework to encourage cross-modally entangled representations while maintaining group-level coherence within a shared latent space. Critically, we introduce a comprehensive combinatorial evaluation framework that systematically assesses representation learners across multiple optimal transport aligners, addressing key limitations in existing evaluation strategies. This framework includes novel simulations that systematically vary shared versus modality-specific perturbation effects enabling principled assessment of method robustness. Our combinatorial benchmarking reveals that there is not yet an aligner that uniformly dominates across settings or modality pairs. Across simulations and two real single-cell genetic perturbation datasets, GROOVE performs on par with or outperforms existing approaches for downstream cross-modal matching and imputation tasks. Our ablation studies demonstrate that GroupCLIP is the key component driving performance gains. These results highlight the importance of leveraging group-level constraints for effective multi-modal representation learning in scenarios where only weak pairing is available.
Joint Embedding vs Reconstruction: Provable Benefits of Latent Space Prediction for Self Supervised Learning
May 18, 2025



Reconstruction and joint embedding have emerged as two leading paradigms in Self Supervised Learning (SSL). Reconstruction methods focus on recovering the original sample from a different view in input space. On the other hand, joint embedding methods align the representations of different views in latent space. Both approaches offer compelling advantages, yet practitioners lack clear guidelines for choosing between them. In this work, we unveil the core mechanisms that distinguish each paradigm. By leveraging closed form solutions for both approaches, we precisely characterize how the view generation process, e.g. data augmentation, impacts the learned representations. We then demonstrate that, unlike supervised learning, both SSL paradigms require a minimal alignment between augmentations and irrelevant features to achieve asymptotic optimality with increasing sample size. Our findings indicate that in scenarios where these irrelevant features have a large magnitude, joint embedding methods are preferable because they impose a strictly weaker alignment condition compared to reconstruction based methods. These results not only clarify the trade offs between the two paradigms but also substantiate the empirical success of joint embedding approaches on real world challenging datasets.
Dynamic Search for Inference-Time Alignment in Diffusion Models
Mar 03, 2025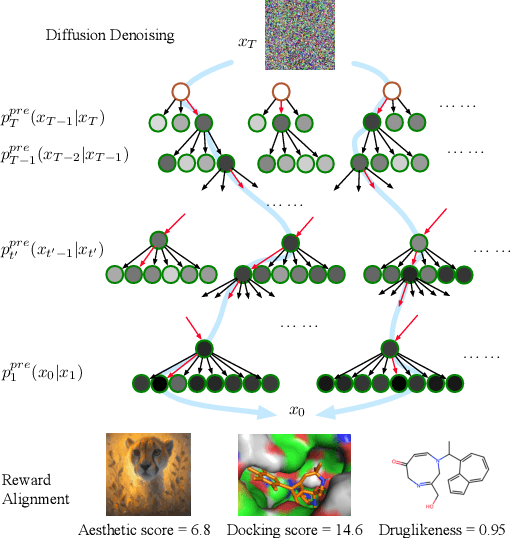
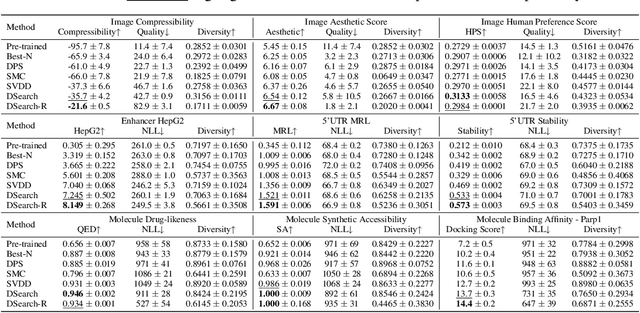
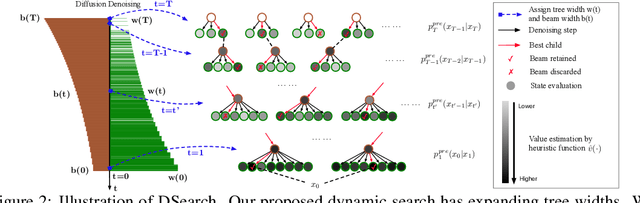

Diffusion models have shown promising generative capabilities across diverse domains, yet aligning their outputs with desired reward functions remains a challenge, particularly in cases where reward functions are non-differentiable. Some gradient-free guidance methods have been developed, but they often struggle to achieve optimal inference-time alignment. In this work, we newly frame inference-time alignment in diffusion as a search problem and propose Dynamic Search for Diffusion (DSearch), which subsamples from denoising processes and approximates intermediate node rewards. It also dynamically adjusts beam width and tree expansion to efficiently explore high-reward generations. To refine intermediate decisions, DSearch incorporates adaptive scheduling based on noise levels and a lookahead heuristic function. We validate DSearch across multiple domains, including biological sequence design, molecular optimization, and image generation, demonstrating superior reward optimization compared to existing approaches.
Reward-Guided Iterative Refinement in Diffusion Models at Test-Time with Applications to Protein and DNA Design
Feb 20, 2025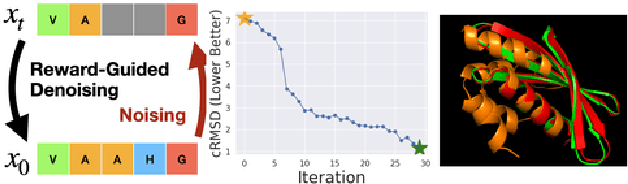
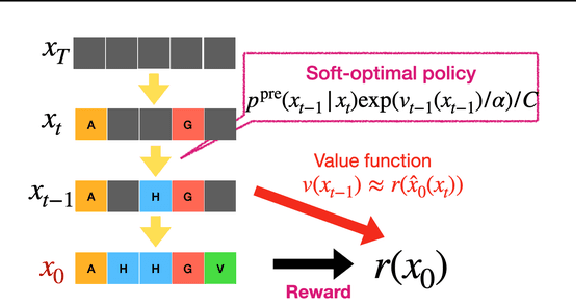
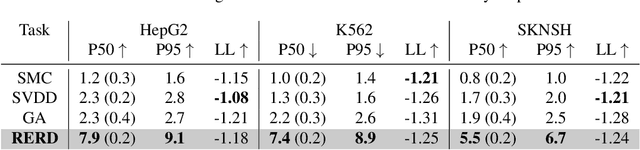
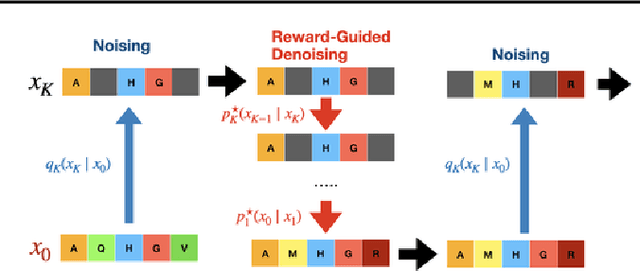
To fully leverage the capabilities of diffusion models, we are often interested in optimizing downstream reward functions during inference. While numerous algorithms for reward-guided generation have been recently proposed due to their significance, current approaches predominantly focus on single-shot generation, transitioning from fully noised to denoised states. We propose a novel framework for inference-time reward optimization with diffusion models inspired by evolutionary algorithms. Our approach employs an iterative refinement process consisting of two steps in each iteration: noising and reward-guided denoising. This sequential refinement allows for the gradual correction of errors introduced during reward optimization. Besides, we provide a theoretical guarantee for our framework. Finally, we demonstrate its superior empirical performance in protein and cell-type-specific regulatory DNA design. The code is available at \href{https://github.com/masa-ue/ProDifEvo-Refinement}{https://github.com/masa-ue/ProDifEvo-Refinement}.
Supervised Contrastive Block Disentanglement
Feb 11, 2025



Real-world datasets often combine data collected under different experimental conditions. This yields larger datasets, but also introduces spurious correlations that make it difficult to model the phenomena of interest. We address this by learning two embeddings to independently represent the phenomena of interest and the spurious correlations. The embedding representing the phenomena of interest is correlated with the target variable $y$, and is invariant to the environment variable $e$. In contrast, the embedding representing the spurious correlations is correlated with $e$. The invariance to $e$ is difficult to achieve on real-world datasets. Our primary contribution is an algorithm called Supervised Contrastive Block Disentanglement (SCBD) that effectively enforces this invariance. It is based purely on Supervised Contrastive Learning, and applies to real-world data better than existing approaches. We empirically validate SCBD on two challenging problems. The first problem is domain generalization, where we achieve strong performance on a synthetic dataset, as well as on Camelyon17-WILDS. We introduce a single hyperparameter $\alpha$ to control the degree of invariance to $e$. When we increase $\alpha$ to strengthen the degree of invariance, out-of-distribution performance improves at the expense of in-distribution performance. The second problem is batch correction, in which we apply SCBD to preserve biological signal and remove inter-well batch effects when modeling single-cell perturbations from 26 million Optical Pooled Screening images.
Reward-Guided Controlled Generation for Inference-Time Alignment in Diffusion Models: Tutorial and Review
Jan 16, 2025



This tutorial provides an in-depth guide on inference-time guidance and alignment methods for optimizing downstream reward functions in diffusion models. While diffusion models are renowned for their generative modeling capabilities, practical applications in fields such as biology often require sample generation that maximizes specific metrics (e.g., stability, affinity in proteins, closeness to target structures). In these scenarios, diffusion models can be adapted not only to generate realistic samples but also to explicitly maximize desired measures at inference time without fine-tuning. This tutorial explores the foundational aspects of such inference-time algorithms. We review these methods from a unified perspective, demonstrating that current techniques -- such as Sequential Monte Carlo (SMC)-based guidance, value-based sampling, and classifier guidance -- aim to approximate soft optimal denoising processes (a.k.a. policies in RL) that combine pre-trained denoising processes with value functions serving as look-ahead functions that predict from intermediate states to terminal rewards. Within this framework, we present several novel algorithms not yet covered in the literature. Furthermore, we discuss (1) fine-tuning methods combined with inference-time techniques, (2) inference-time algorithms based on search algorithms such as Monte Carlo tree search, which have received limited attention in current research, and (3) connections between inference-time algorithms in language models and diffusion models. The code of this tutorial on protein design is available at https://github.com/masa-ue/AlignInversePro
Fine-Tuning Discrete Diffusion Models via Reward Optimization with Applications to DNA and Protein Design
Oct 17, 2024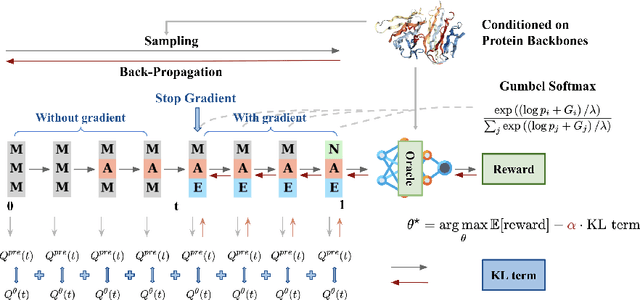
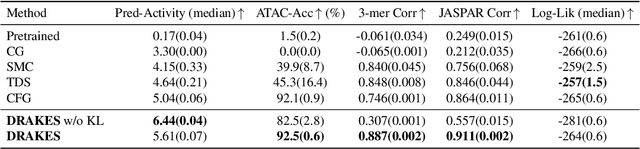
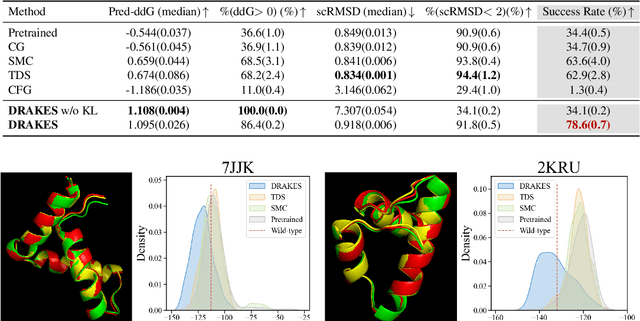

Recent studies have demonstrated the strong empirical performance of diffusion models on discrete sequences across domains from natural language to biological sequence generation. For example, in the protein inverse folding task, conditional diffusion models have achieved impressive results in generating natural-like sequences that fold back into the original structure. However, practical design tasks often require not only modeling a conditional distribution but also optimizing specific task objectives. For instance, we may prefer protein sequences with high stability. To address this, we consider the scenario where we have pre-trained discrete diffusion models that can generate natural-like sequences, as well as reward models that map sequences to task objectives. We then formulate the reward maximization problem within discrete diffusion models, analogous to reinforcement learning (RL), while minimizing the KL divergence against pretrained diffusion models to preserve naturalness. To solve this RL problem, we propose a novel algorithm, DRAKES, that enables direct backpropagation of rewards through entire trajectories generated by diffusion models, by making the originally non-differentiable trajectories differentiable using the Gumbel-Softmax trick. Our theoretical analysis indicates that our approach can generate sequences that are both natural-like and yield high rewards. While similar tasks have been recently explored in diffusion models for continuous domains, our work addresses unique algorithmic and theoretical challenges specific to discrete diffusion models, which arise from their foundation in continuous-time Markov chains rather than Brownian motion. Finally, we demonstrate the effectiveness of DRAKES in generating DNA and protein sequences that optimize enhancer activity and protein stability, respectively, important tasks for gene therapies and protein-based therapeutics.
How to Build the Virtual Cell with Artificial Intelligence: Priorities and Opportunities
Sep 18, 2024

The cell is arguably the smallest unit of life and is central to understanding biology. Accurate modeling of cells is important for this understanding as well as for determining the root causes of disease. Recent advances in artificial intelligence (AI), combined with the ability to generate large-scale experimental data, present novel opportunities to model cells. Here we propose a vision of AI-powered Virtual Cells, where robust representations of cells and cellular systems under different conditions are directly learned from growing biological data across measurements and scales. We discuss desired capabilities of AI Virtual Cells, including generating universal representations of biological entities across scales, and facilitating interpretable in silico experiments to predict and understand their behavior using Virtual Instruments. We further address the challenges, opportunities and requirements to realize this vision including data needs, evaluation strategies, and community standards and engagement to ensure biological accuracy and broad utility. We envision a future where AI Virtual Cells help identify new drug targets, predict cellular responses to perturbations, as well as scale hypothesis exploration. With open science collaborations across the biomedical ecosystem that includes academia, philanthropy, and the biopharma and AI industries, a comprehensive predictive understanding of cell mechanisms and interactions is within reach.