 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRAG-Enhanced Collaborative LLM Agents for Drug Discovery
Feb 22, 2025Recent advances in large language models (LLMs) have shown great potential to accelerate drug discovery. However, the specialized nature of biochemical data often necessitates costly domain-specific fine-tuning, posing critical challenges. First, it hinders the application of more flexible general-purpose LLMs in cutting-edge drug discovery tasks. More importantly, it impedes the rapid integration of the vast amounts of scientific data continuously generated through experiments and research. To investigate these challenges, we propose CLADD, a retrieval-augmented generation (RAG)-empowered agentic system tailored to drug discovery tasks. Through the collaboration of multiple LLM agents, CLADD dynamically retrieves information from biomedical knowledge bases, contextualizes query molecules, and integrates relevant evidence to generate responses -- all without the need for domain-specific fine-tuning. Crucially, we tackle key obstacles in applying RAG workflows to biochemical data, including data heterogeneity, ambiguity, and multi-source integration. We demonstrate the flexibility and effectiveness of this framework across a variety of drug discovery tasks, showing that it outperforms general-purpose and domain-specific LLMs as well as traditional deep learning approaches.
MolCap-Arena: A Comprehensive Captioning Benchmark on Language-Enhanced Molecular Property Prediction
Nov 01, 2024



Bridging biomolecular modeling with natural language information, particularly through large language models (LLMs), has recently emerged as a promising interdisciplinary research area. LLMs, having been trained on large corpora of scientific documents, demonstrate significant potential in understanding and reasoning about biomolecules by providing enriched contextual and domain knowledge. However, the extent to which LLM-driven insights can improve performance on complex predictive tasks (e.g., toxicity) remains unclear. Further, the extent to which relevant knowledge can be extracted from LLMs also remains unknown. In this study, we present Molecule Caption Arena: the first comprehensive benchmark of LLM-augmented molecular property prediction. We evaluate over twenty LLMs, including both general-purpose and domain-specific molecule captioners, across diverse prediction tasks. To this goal, we introduce a novel, battle-based rating system. Our findings confirm the ability of LLM-extracted knowledge to enhance state-of-the-art molecular representations, with notable model-, prompt-, and dataset-specific variations. Code, resources, and data are available at github.com/Genentech/molcap-arena.
Cell Morphology-Guided Small Molecule Generation with GFlowNets
Aug 09, 2024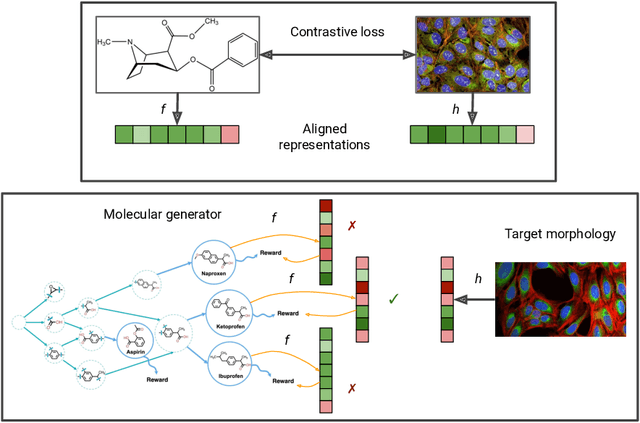

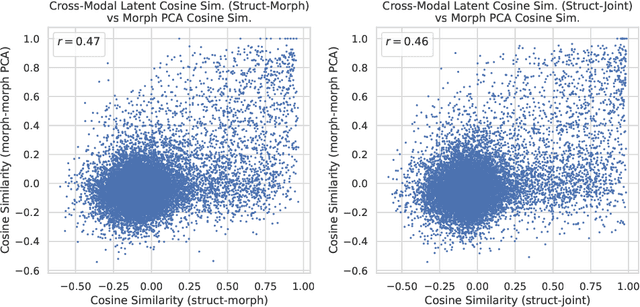

High-content phenotypic screening, including high-content imaging (HCI), has gained popularity in the last few years for its ability to characterize novel therapeutics without prior knowledge of the protein target. When combined with deep learning techniques to predict and represent molecular-phenotype interactions, these advancements hold the potential to significantly accelerate and enhance drug discovery applications. This work focuses on the novel task of HCI-guided molecular design. Generative models for molecule design could be guided by HCI data, for example with a supervised model that links molecules to phenotypes of interest as a reward function. However, limited labeled data, combined with the high-dimensional readouts, can make training these methods challenging and impractical. We consider an alternative approach in which we leverage an unsupervised multimodal joint embedding to define a latent similarity as a reward for GFlowNets. The proposed model learns to generate new molecules that could produce phenotypic effects similar to those of the given image target, without relying on pre-annotated phenotypic labels. We demonstrate that the proposed method generates molecules with high morphological and structural similarity to the target, increasing the likelihood of similar biological activity, as confirmed by an independent oracle model.
Adding Conditional Control to Diffusion Models with Reinforcement Learning
Jun 17, 2024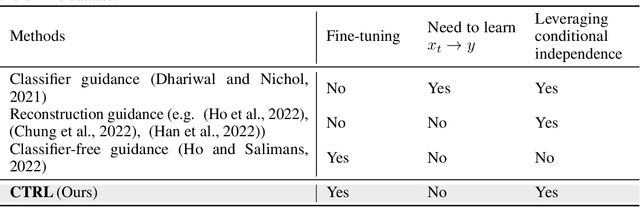
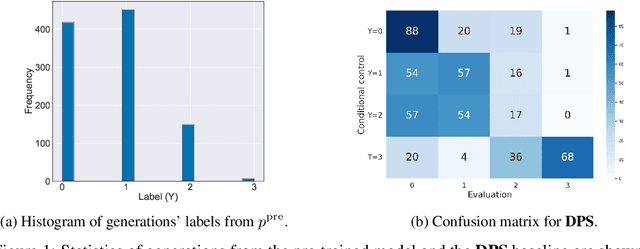
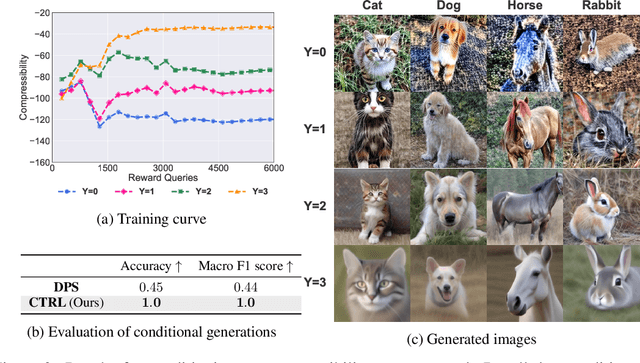
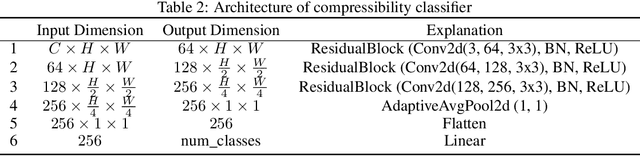
Diffusion models are powerful generative models that allow for precise control over the characteristics of the generated samples. While these diffusion models trained on large datasets have achieved success, there is often a need to introduce additional controls in downstream fine-tuning processes, treating these powerful models as pre-trained diffusion models. This work presents a novel method based on reinforcement learning (RL) to add additional controls, leveraging an offline dataset comprising inputs and corresponding labels. We formulate this task as an RL problem, with the classifier learned from the offline dataset and the KL divergence against pre-trained models serving as the reward functions. We introduce our method, $\textbf{CTRL}$ ($\textbf{C}$onditioning pre-$\textbf{T}$rained diffusion models with $\textbf{R}$einforcement $\textbf{L}$earning), which produces soft-optimal policies that maximize the abovementioned reward functions. We formally demonstrate that our method enables sampling from the conditional distribution conditioned on additional controls during inference. Our RL-based approach offers several advantages over existing methods. Compared to commonly used classifier-free guidance, our approach improves sample efficiency, and can greatly simplify offline dataset construction by exploiting conditional independence between the inputs and additional controls. Furthermore, unlike classifier guidance, we avoid the need to train classifiers from intermediate states to additional controls.
Bridging Model-Based Optimization and Generative Modeling via Conservative Fine-Tuning of Diffusion Models
May 31, 2024



AI-driven design problems, such as DNA/protein sequence design, are commonly tackled from two angles: generative modeling, which efficiently captures the feasible design space (e.g., natural images or biological sequences), and model-based optimization, which utilizes reward models for extrapolation. To combine the strengths of both approaches, we adopt a hybrid method that fine-tunes cutting-edge diffusion models by optimizing reward models through RL. Although prior work has explored similar avenues, they primarily focus on scenarios where accurate reward models are accessible. In contrast, we concentrate on an offline setting where a reward model is unknown, and we must learn from static offline datasets, a common scenario in scientific domains. In offline scenarios, existing approaches tend to suffer from overoptimization, as they may be misled by the reward model in out-of-distribution regions. To address this, we introduce a conservative fine-tuning approach, BRAID, by optimizing a conservative reward model, which includes additional penalization outside of offline data distributions. Through empirical and theoretical analysis, we demonstrate the capability of our approach to outperform the best designs in offline data, leveraging the extrapolation capabilities of reward models while avoiding the generation of invalid designs through pre-trained diffusion models.
Fine-Tuning of Continuous-Time Diffusion Models as Entropy-Regularized Control
Feb 28, 2024



Diffusion models excel at capturing complex data distributions, such as those of natural images and proteins. While diffusion models are trained to represent the distribution in the training dataset, we often are more concerned with other properties, such as the aesthetic quality of the generated images or the functional properties of generated proteins. Diffusion models can be finetuned in a goal-directed way by maximizing the value of some reward function (e.g., the aesthetic quality of an image). However, these approaches may lead to reduced sample diversity, significant deviations from the training data distribution, and even poor sample quality due to the exploitation of an imperfect reward function. The last issue often occurs when the reward function is a learned model meant to approximate a ground-truth "genuine" reward, as is the case in many practical applications. These challenges, collectively termed "reward collapse," pose a substantial obstacle. To address this reward collapse, we frame the finetuning problem as entropy-regularized control against the pretrained diffusion model, i.e., directly optimizing entropy-enhanced rewards with neural SDEs. We present theoretical and empirical evidence that demonstrates our framework is capable of efficiently generating diverse samples with high genuine rewards, mitigating the overoptimization of imperfect reward models.
Feedback Efficient Online Fine-Tuning of Diffusion Models
Feb 27, 2024



Diffusion models excel at modeling complex data distributions, including those of images, proteins, and small molecules. However, in many cases, our goal is to model parts of the distribution that maximize certain properties: for example, we may want to generate images with high aesthetic quality, or molecules with high bioactivity. It is natural to frame this as a reinforcement learning (RL) problem, in which the objective is to fine-tune a diffusion model to maximize a reward function that corresponds to some property. Even with access to online queries of the ground-truth reward function, efficiently discovering high-reward samples can be challenging: they might have a low probability in the initial distribution, and there might be many infeasible samples that do not even have a well-defined reward (e.g., unnatural images or physically impossible molecules). In this work, we propose a novel reinforcement learning procedure that efficiently explores on the manifold of feasible samples. We present a theoretical analysis providing a regret guarantee, as well as empirical validation across three domains: images, biological sequences, and molecules.
Toward the Identifiability of Comparative Deep Generative Models
Jan 29, 2024



Deep Generative Models (DGMs) are versatile tools for learning data representations while adequately incorporating domain knowledge such as the specification of conditional probability distributions. Recently proposed DGMs tackle the important task of comparing data sets from different sources. One such example is the setting of contrastive analysis that focuses on describing patterns that are enriched in a target data set compared to a background data set. The practical deployment of those models often assumes that DGMs naturally infer interpretable and modular latent representations, which is known to be an issue in practice. Consequently, existing methods often rely on ad-hoc regularization schemes, although without any theoretical grounding. Here, we propose a theory of identifiability for comparative DGMs by extending recent advances in the field of non-linear independent component analysis. We show that, while these models lack identifiability across a general class of mixing functions, they surprisingly become identifiable when the mixing function is piece-wise affine (e.g., parameterized by a ReLU neural network). We also investigate the impact of model misspecification, and empirically show that previously proposed regularization techniques for fitting comparative DGMs help with identifiability when the number of latent variables is not known in advance. Finally, we introduce a novel methodology for fitting comparative DGMs that improves the treatment of multiple data sources via multi-objective optimization and that helps adjust the hyperparameter for the regularization in an interpretable manner, using constrained optimization. We empirically validate our theory and new methodology using simulated data as well as a recent data set of genetic perturbations in cells profiled via single-cell RNA sequencing.
* 45 pages, 3 figures
Conformalized Deep Splines for Optimal and Efficient Prediction Sets
Nov 01, 2023


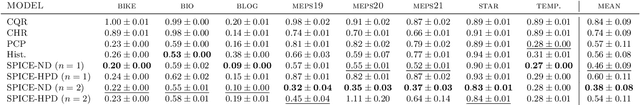
Uncertainty estimation is critical in high-stakes machine learning applications. One effective way to estimate uncertainty is conformal prediction, which can provide predictive inference with statistical coverage guarantees. We present a new conformal regression method, Spline Prediction Intervals via Conformal Estimation (SPICE), that estimates the conditional density using neural-network-parameterized splines. We prove universal approximation and optimality results for SPICE, which are empirically validated by our experiments. SPICE is compatible with two different efficient-to-compute conformal scores, one oracle-optimal for marginal coverage (SPICE-ND) and the other asymptotically optimal for conditional coverage (SPICE-HPD). Results on benchmark datasets demonstrate SPICE-ND models achieve the smallest average prediction set sizes, including average size reductions of nearly 50% for some datasets compared to the next best baseline. SPICE-HPD models achieve the best conditional coverage compared to baselines. The SPICE implementation is made available.
Towards Understanding and Improving GFlowNet Training
May 11, 2023


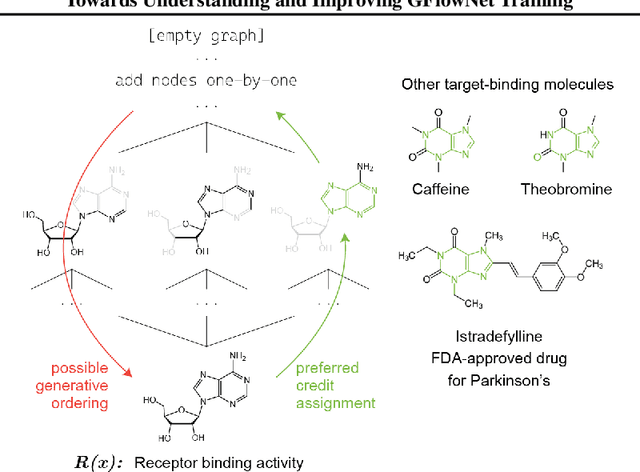
Generative flow networks (GFlowNets) are a family of algorithms that learn a generative policy to sample discrete objects $x$ with non-negative reward $R(x)$. Learning objectives guarantee the GFlowNet samples $x$ from the target distribution $p^*(x) \propto R(x)$ when loss is globally minimized over all states or trajectories, but it is unclear how well they perform with practical limits on training resources. We introduce an efficient evaluation strategy to compare the learned sampling distribution to the target reward distribution. As flows can be underdetermined given training data, we clarify the importance of learned flows to generalization and matching $p^*(x)$ in practice. We investigate how to learn better flows, and propose (i) prioritized replay training of high-reward $x$, (ii) relative edge flow policy parametrization, and (iii) a novel guided trajectory balance objective, and show how it can solve a substructure credit assignment problem. We substantially improve sample efficiency on biochemical design tasks.