 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSynCoGen: Synthesizable 3D Molecule Generation via Joint Reaction and Coordinate Modeling
Jul 16, 2025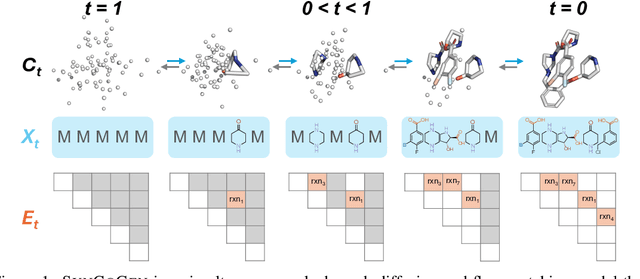
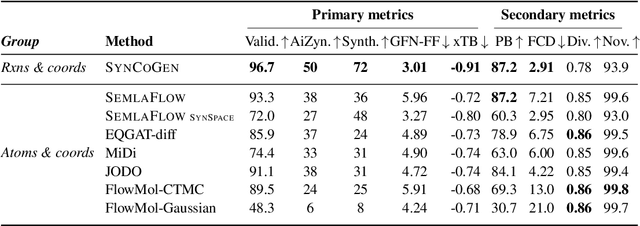
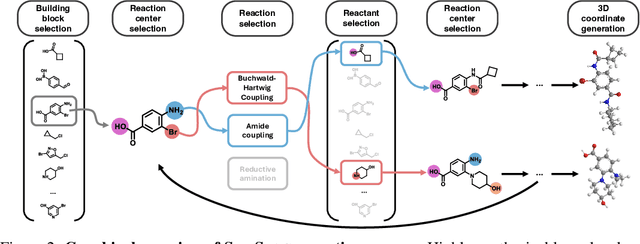
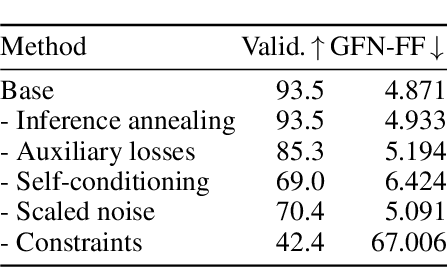
Ensuring synthesizability in generative small molecule design remains a major challenge. While recent developments in synthesizable molecule generation have demonstrated promising results, these efforts have been largely confined to 2D molecular graph representations, limiting the ability to perform geometry-based conditional generation. In this work, we present SynCoGen (Synthesizable Co-Generation), a single framework that combines simultaneous masked graph diffusion and flow matching for synthesizable 3D molecule generation. SynCoGen samples from the joint distribution of molecular building blocks, chemical reactions, and atomic coordinates. To train the model, we curated SynSpace, a dataset containing over 600K synthesis-aware building block graphs and 3.3M conformers. SynCoGen achieves state-of-the-art performance in unconditional small molecule graph and conformer generation, and the model delivers competitive performance in zero-shot molecular linker design for protein ligand generation in drug discovery. Overall, this multimodal formulation represents a foundation for future applications enabled by non-autoregressive molecular generation, including analog expansion, lead optimization, and direct structure conditioning.
Measuring Scientific Capabilities of Language Models with a Systems Biology Dry Lab
Jul 02, 2025
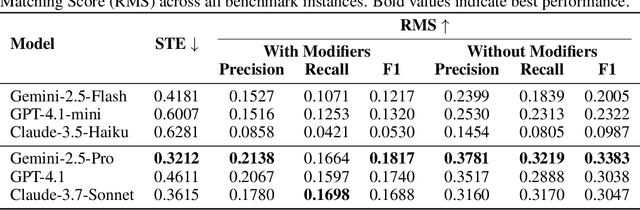
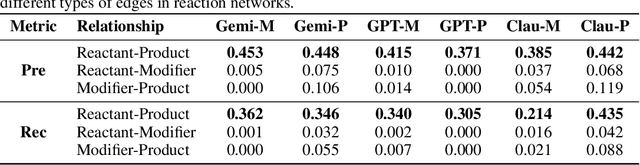
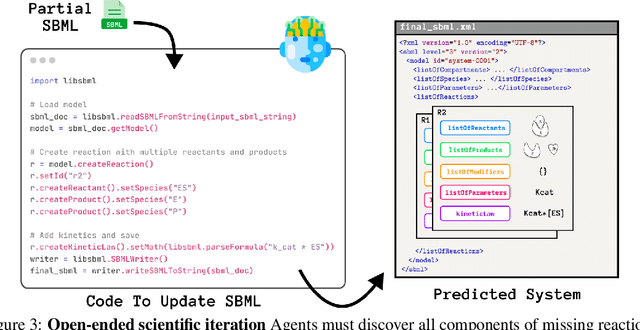
Designing experiments and result interpretations are core scientific competencies, particularly in biology, where researchers perturb complex systems to uncover the underlying systems. Recent efforts to evaluate the scientific capabilities of large language models (LLMs) fail to test these competencies because wet-lab experimentation is prohibitively expensive: in expertise, time and equipment. We introduce SciGym, a first-in-class benchmark that assesses LLMs' iterative experiment design and analysis abilities in open-ended scientific discovery tasks. SciGym overcomes the challenge of wet-lab costs by running a dry lab of biological systems. These models, encoded in Systems Biology Markup Language, are efficient for generating simulated data, making them ideal testbeds for experimentation on realistically complex systems. We evaluated six frontier LLMs on 137 small systems, and released a total of 350 systems. Our evaluation shows that while more capable models demonstrated superior performance, all models' performance declined significantly as system complexity increased, suggesting substantial room for improvement in the scientific capabilities of LLM agents.
Learning Decision Trees as Amortized Structure Inference
Mar 10, 2025Building predictive models for tabular data presents fundamental challenges, notably in scaling consistently, i.e., more resources translating to better performance, and generalizing systematically beyond the training data distribution. Designing decision tree models remains especially challenging given the intractably large search space, and most existing methods rely on greedy heuristics, while deep learning inductive biases expect a temporal or spatial structure not naturally present in tabular data. We propose a hybrid amortized structure inference approach to learn predictive decision tree ensembles given data, formulating decision tree construction as a sequential planning problem. We train a deep reinforcement learning (GFlowNet) policy to solve this problem, yielding a generative model that samples decision trees from the Bayesian posterior. We show that our approach, DT-GFN, outperforms state-of-the-art decision tree and deep learning methods on standard classification benchmarks derived from real-world data, robustness to distribution shifts, and anomaly detection, all while yielding interpretable models with shorter description lengths. Samples from the trained DT-GFN model can be ensembled to construct a random forest, and we further show that the performance of scales consistently in ensemble size, yielding ensembles of predictors that continue to generalize systematically.
Action abstractions for amortized sampling
Oct 19, 2024As trajectories sampled by policies used by reinforcement learning (RL) and generative flow networks (GFlowNets) grow longer, credit assignment and exploration become more challenging, and the long planning horizon hinders mode discovery and generalization. The challenge is particularly pronounced in entropy-seeking RL methods, such as generative flow networks, where the agent must learn to sample from a structured distribution and discover multiple high-reward states, each of which take many steps to reach. To tackle this challenge, we propose an approach to incorporate the discovery of action abstractions, or high-level actions, into the policy optimization process. Our approach involves iteratively extracting action subsequences commonly used across many high-reward trajectories and `chunking' them into a single action that is added to the action space. In empirical evaluation on synthetic and real-world environments, our approach demonstrates improved sample efficiency performance in discovering diverse high-reward objects, especially on harder exploration problems. We also observe that the abstracted high-order actions are interpretable, capturing the latent structure of the reward landscape of the action space. This work provides a cognitively motivated approach to action abstraction in RL and is the first demonstration of hierarchical planning in amortized sequential sampling.
Cell Morphology-Guided Small Molecule Generation with GFlowNets
Aug 09, 2024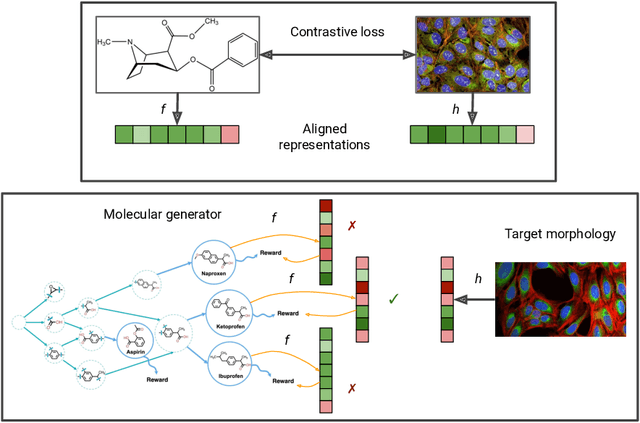

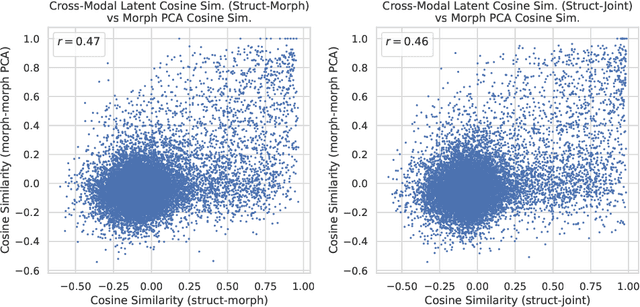

High-content phenotypic screening, including high-content imaging (HCI), has gained popularity in the last few years for its ability to characterize novel therapeutics without prior knowledge of the protein target. When combined with deep learning techniques to predict and represent molecular-phenotype interactions, these advancements hold the potential to significantly accelerate and enhance drug discovery applications. This work focuses on the novel task of HCI-guided molecular design. Generative models for molecule design could be guided by HCI data, for example with a supervised model that links molecules to phenotypes of interest as a reward function. However, limited labeled data, combined with the high-dimensional readouts, can make training these methods challenging and impractical. We consider an alternative approach in which we leverage an unsupervised multimodal joint embedding to define a latent similarity as a reward for GFlowNets. The proposed model learns to generate new molecules that could produce phenotypic effects similar to those of the given image target, without relying on pre-annotated phenotypic labels. We demonstrate that the proposed method generates molecules with high morphological and structural similarity to the target, increasing the likelihood of similar biological activity, as confirmed by an independent oracle model.
RetroGFN: Diverse and Feasible Retrosynthesis using GFlowNets
Jun 26, 2024Single-step retrosynthesis aims to predict a set of reactions that lead to the creation of a target molecule, which is a crucial task in molecular discovery. Although a target molecule can often be synthesized with multiple different reactions, it is not clear how to verify the feasibility of a reaction, because the available datasets cover only a tiny fraction of the possible solutions. Consequently, the existing models are not encouraged to explore the space of possible reactions sufficiently. In this paper, we propose a novel single-step retrosynthesis model, RetroGFN, that can explore outside the limited dataset and return a diverse set of feasible reactions by leveraging a feasibility proxy model during the training. We show that RetroGFN achieves competitive results on standard top-k accuracy while outperforming existing methods on round-trip accuracy. Moreover, we provide empirical arguments in favor of using round-trip accuracy which expands the notion of feasibility with respect to the standard top-k accuracy metric.
Towards DNA-Encoded Library Generation with GFlowNets
Apr 15, 2024DNA-encoded libraries (DELs) are a powerful approach for rapidly screening large numbers of diverse compounds. One of the key challenges in using DELs is library design, which involves choosing the building blocks that will be combinatorially combined to produce the final library. In this paper we consider the task of protein-protein interaction (PPI) biased DEL design. To this end, we evaluate several machine learning algorithms on the PPI modulation task and use them as a reward for the proposed GFlowNet-based generative approach. We additionally investigate the possibility of using structural information about building blocks to design a hierarchical action space for the GFlowNet. The observed results indicate that GFlowNets are a promising approach for generating diverse combinatorial library candidates.
Towards equilibrium molecular conformation generation with GFlowNets
Oct 20, 2023



Sampling diverse, thermodynamically feasible molecular conformations plays a crucial role in predicting properties of a molecule. In this paper we propose to use GFlowNet for sampling conformations of small molecules from the Boltzmann distribution, as determined by the molecule's energy. The proposed approach can be used in combination with energy estimation methods of different fidelity and discovers a diverse set of low-energy conformations for highly flexible drug-like molecules. We demonstrate that GFlowNet can reproduce molecular potential energy surfaces by sampling proportionally to the Boltzmann distribution.
Towards Foundational Models for Molecular Learning on Large-Scale Multi-Task Datasets
Oct 18, 2023



Recently, pre-trained foundation models have enabled significant advancements in multiple fields. In molecular machine learning, however, where datasets are often hand-curated, and hence typically small, the lack of datasets with labeled features, and codebases to manage those datasets, has hindered the development of foundation models. In this work, we present seven novel datasets categorized by size into three distinct categories: ToyMix, LargeMix and UltraLarge. These datasets push the boundaries in both the scale and the diversity of supervised labels for molecular learning. They cover nearly 100 million molecules and over 3000 sparsely defined tasks, totaling more than 13 billion individual labels of both quantum and biological nature. In comparison, our datasets contain 300 times more data points than the widely used OGB-LSC PCQM4Mv2 dataset, and 13 times more than the quantum-only QM1B dataset. In addition, to support the development of foundational models based on our proposed datasets, we present the Graphium graph machine learning library which simplifies the process of building and training molecular machine learning models for multi-task and multi-level molecular datasets. Finally, we present a range of baseline results as a starting point of multi-task and multi-level training on these datasets. Empirically, we observe that performance on low-resource biological datasets show improvement by also training on large amounts of quantum data. This indicates that there may be potential in multi-task and multi-level training of a foundation model and fine-tuning it to resource-constrained downstream tasks.
Crystal-GFN: sampling crystals with desirable properties and constraints
Oct 07, 2023Accelerating material discovery holds the potential to greatly help mitigate the climate crisis. Discovering new solid-state crystals such as electrocatalysts, ionic conductors or photovoltaics can have a crucial impact, for instance, in improving the efficiency of renewable energy production and storage. In this paper, we introduce Crystal-GFlowNet, a generative model of crystal structures that sequentially samples a crystal's composition, space group and lattice parameters. This domain-inspired approach enables the flexible incorporation of physical and geometrical constraints, as well as the use of any available predictive model of a desired property as an objective function. We evaluate the capabilities of Crystal-GFlowNet by using as objective the formation energy of a crystal structure, as predicted by a new proxy model trained on MatBench. The results demonstrate that Crystal-GFlowNet is able to sample diverse crystals with low formation energy.