 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTowards Autonomous Mechanistic Reasoning in Virtual Cells
Apr 14, 2026Large language models (LLMs) have recently gained significant attention as a promising approach to accelerate scientific discovery. However, their application in open-ended scientific domains such as biology remains limited, primarily due to the lack of factually grounded and actionable explanations. To address this, we introduce a structured explanation formalism for virtual cells that represents biological reasoning as mechanistic action graphs, enabling systematic verification and falsification. Building upon this, we propose VCR-Agent, a multi-agent framework that integrates biologically grounded knowledge retrieval with a verifier-based filtering approach to generate and validate mechanistic reasoning autonomously. Using this framework, we release VC-TRACES dataset, which consists of verified mechanistic explanations derived from the Tahoe-100M atlas. Empirically, we demonstrate that training with these explanations improves factual precision and provides a more effective supervision signal for downstream gene expression prediction. These results underscore the importance of reliable mechanistic reasoning for virtual cells, achieved through the synergy of multi-agent and rigorous verification.
Amortized Sampling with Transferable Normalizing Flows
Aug 25, 2025Efficient equilibrium sampling of molecular conformations remains a core challenge in computational chemistry and statistical inference. Classical approaches such as molecular dynamics or Markov chain Monte Carlo inherently lack amortization; the computational cost of sampling must be paid in-full for each system of interest. The widespread success of generative models has inspired interest into overcoming this limitation through learning sampling algorithms. Despite performing on par with conventional methods when trained on a single system, learned samplers have so far demonstrated limited ability to transfer across systems. We prove that deep learning enables the design of scalable and transferable samplers by introducing Prose, a 280 million parameter all-atom transferable normalizing flow trained on a corpus of peptide molecular dynamics trajectories up to 8 residues in length. Prose draws zero-shot uncorrelated proposal samples for arbitrary peptide systems, achieving the previously intractable transferability across sequence length, whilst retaining the efficient likelihood evaluation of normalizing flows. Through extensive empirical evaluation we demonstrate the efficacy of Prose as a proposal for a variety of sampling algorithms, finding a simple importance sampling-based finetuning procedure to achieve superior performance to established methods such as sequential Monte Carlo on unseen tetrapeptides. We open-source the Prose codebase, model weights, and training dataset, to further stimulate research into amortized sampling methods and finetuning objectives.
Virtual Cells: Predict, Explain, Discover
May 20, 2025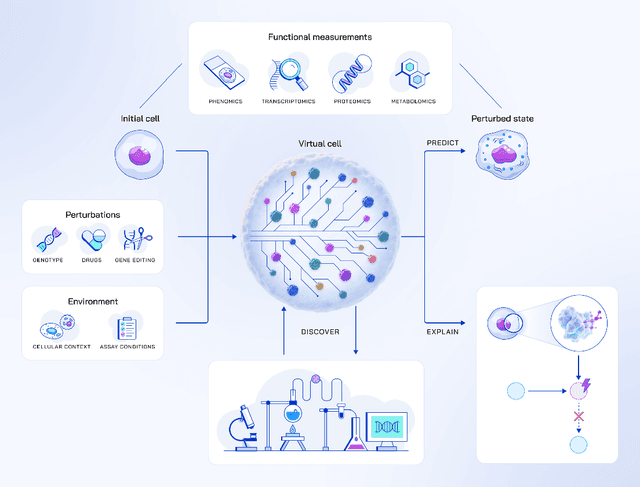



Drug discovery is fundamentally a process of inferring the effects of treatments on patients, and would therefore benefit immensely from computational models that can reliably simulate patient responses, enabling researchers to generate and test large numbers of therapeutic hypotheses safely and economically before initiating costly clinical trials. Even a more specific model that predicts the functional response of cells to a wide range of perturbations would be tremendously valuable for discovering safe and effective treatments that successfully translate to the clinic. Creating such virtual cells has long been a goal of the computational research community that unfortunately remains unachieved given the daunting complexity and scale of cellular biology. Nevertheless, recent advances in AI, computing power, lab automation, and high-throughput cellular profiling provide new opportunities for reaching this goal. In this perspective, we present a vision for developing and evaluating virtual cells that builds on our experience at Recursion. We argue that in order to be a useful tool to discover novel biology, virtual cells must accurately predict the functional response of a cell to perturbations and explain how the predicted response is a consequence of modifications to key biomolecular interactions. We then introduce key principles for designing therapeutically-relevant virtual cells, describe a lab-in-the-loop approach for generating novel insights with them, and advocate for biologically-grounded benchmarks to guide virtual cell development. Finally, we make the case that our approach to virtual cells provides a useful framework for building other models at higher levels of organization, including virtual patients. We hope that these directions prove useful to the research community in developing virtual models optimized for positive impact on drug discovery outcomes.
OpenQDC: Open Quantum Data Commons
Nov 29, 2024



Machine Learning Interatomic Potentials (MLIPs) are a highly promising alternative to force-fields for molecular dynamics (MD) simulations, offering precise and rapid energy and force calculations. However, Quantum-Mechanical (QM) datasets, crucial for MLIPs, are fragmented across various repositories, hindering accessibility and model development. We introduce the openQDC package, consolidating 37 QM datasets from over 250 quantum methods and 400 million geometries into a single, accessible resource. These datasets are meticulously preprocessed, and standardized for MLIP training, covering a wide range of chemical elements and interactions relevant in organic chemistry. OpenQDC includes tools for normalization and integration, easily accessible via Python. Experiments with well-known architectures like SchNet, TorchMD-Net, and DimeNet reveal challenges for those architectures and constitute a leaderboard to accelerate benchmarking and guide novel algorithms development. Continuously adding datasets to OpenQDC will democratize QM dataset access, foster more collaboration and innovation, enhance MLIP development, and support their adoption in the MD field.
ET-Flow: Equivariant Flow-Matching for Molecular Conformer Generation
Oct 29, 2024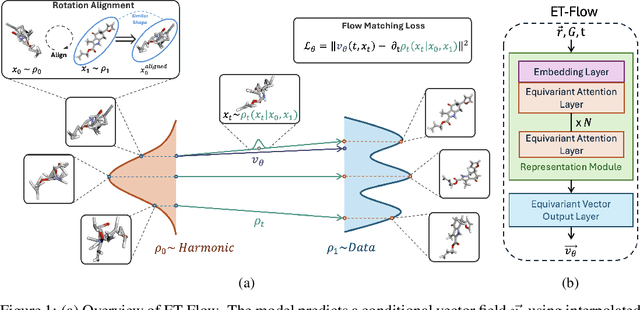
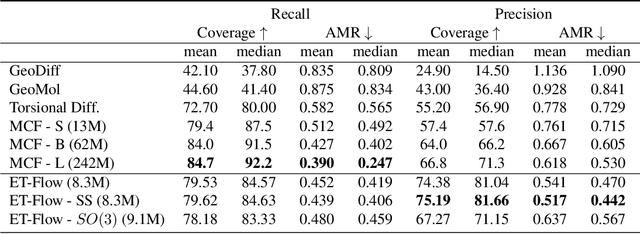
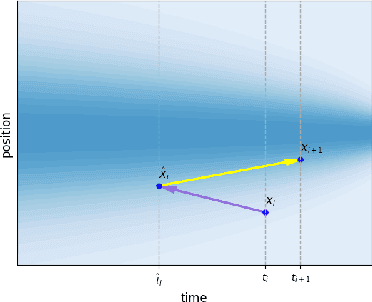
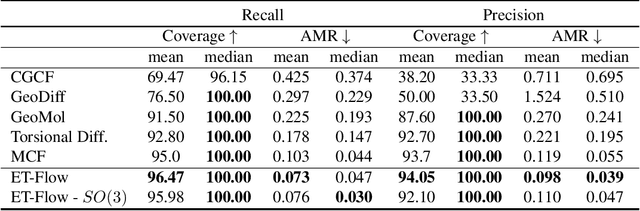
Predicting low-energy molecular conformations given a molecular graph is an important but challenging task in computational drug discovery. Existing state-of-the-art approaches either resort to large scale transformer-based models that diffuse over conformer fields, or use computationally expensive methods to generate initial structures and diffuse over torsion angles. In this work, we introduce Equivariant Transformer Flow (ET-Flow). We showcase that a well-designed flow matching approach with equivariance and harmonic prior alleviates the need for complex internal geometry calculations and large architectures, contrary to the prevailing methods in the field. Our approach results in a straightforward and scalable method that directly operates on all-atom coordinates with minimal assumptions. With the advantages of equivariance and flow matching, ET-Flow significantly increases the precision and physical validity of the generated conformers, while being a lighter model and faster at inference. Code is available https://github.com/shenoynikhil/ETFlow.
On the Scalability of GNNs for Molecular Graphs
Apr 17, 2024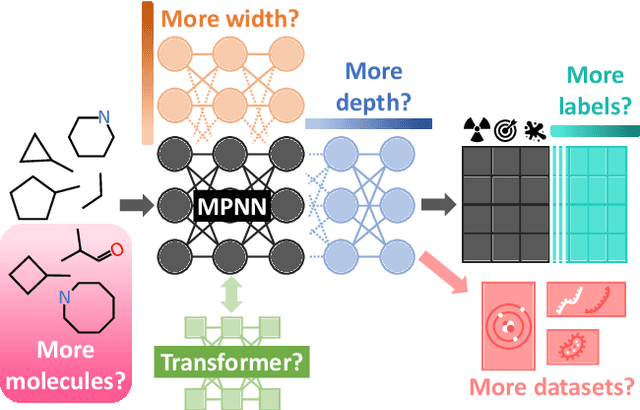

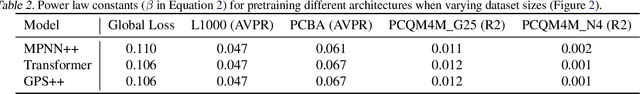
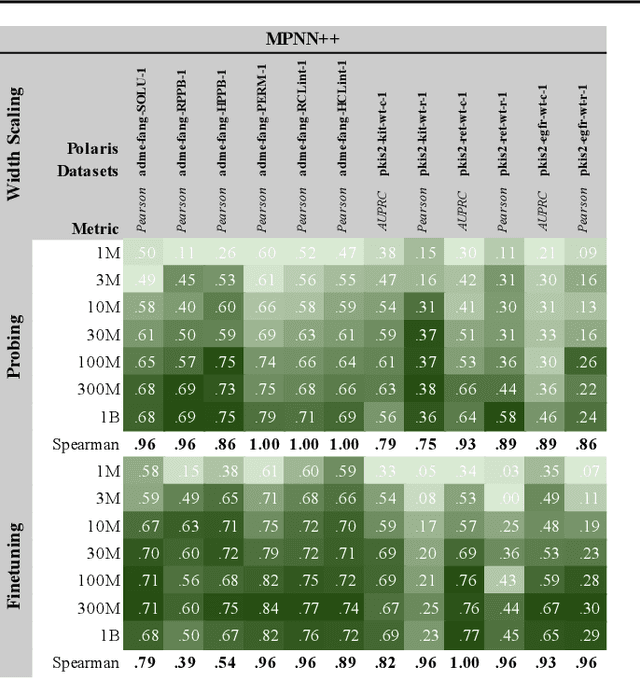
Scaling deep learning models has been at the heart of recent revolutions in language modelling and image generation. Practitioners have observed a strong relationship between model size, dataset size, and performance. However, structure-based architectures such as Graph Neural Networks (GNNs) are yet to show the benefits of scale mainly due to the lower efficiency of sparse operations, large data requirements, and lack of clarity about the effectiveness of various architectures. We address this drawback of GNNs by studying their scaling behavior. Specifically, we analyze message-passing networks, graph Transformers, and hybrid architectures on the largest public collection of 2D molecular graphs. For the first time, we observe that GNNs benefit tremendously from the increasing scale of depth, width, number of molecules, number of labels, and the diversity in the pretraining datasets, resulting in a 30.25% improvement when scaling to 1 billion parameters and 28.98% improvement when increasing size of dataset to eightfold. We further demonstrate strong finetuning scaling behavior on 38 tasks, outclassing previous large models. We hope that our work paves the way for an era where foundational GNNs drive pharmaceutical drug discovery.
Masked Autoencoders for Microscopy are Scalable Learners of Cellular Biology
Apr 16, 2024



Featurizing microscopy images for use in biological research remains a significant challenge, especially for large-scale experiments spanning millions of images. This work explores the scaling properties of weakly supervised classifiers and self-supervised masked autoencoders (MAEs) when training with increasingly larger model backbones and microscopy datasets. Our results show that ViT-based MAEs outperform weakly supervised classifiers on a variety of tasks, achieving as much as a 11.5% relative improvement when recalling known biological relationships curated from public databases. Additionally, we develop a new channel-agnostic MAE architecture (CA-MAE) that allows for inputting images of different numbers and orders of channels at inference time. We demonstrate that CA-MAEs effectively generalize by inferring and evaluating on a microscopy image dataset (JUMP-CP) generated under different experimental conditions with a different channel structure than our pretraining data (RPI-93M). Our findings motivate continued research into scaling self-supervised learning on microscopy data in order to create powerful foundation models of cellular biology that have the potential to catalyze advancements in drug discovery and beyond.
Generating QM1B with PySCF$_{\text{IPU}}$
Nov 02, 2023The emergence of foundation models in Computer Vision and Natural Language Processing have resulted in immense progress on downstream tasks. This progress was enabled by datasets with billions of training examples. Similar benefits are yet to be unlocked for quantum chemistry, where the potential of deep learning is constrained by comparatively small datasets with 100k to 20M training examples. These datasets are limited in size because the labels are computed using the accurate (but computationally demanding) predictions of Density Functional Theory (DFT). Notably, prior DFT datasets were created using CPU supercomputers without leveraging hardware acceleration. In this paper, we take a first step towards utilising hardware accelerators by introducing the data generator PySCF$_{\text{IPU}}$ using Intelligence Processing Units (IPUs). This allowed us to create the dataset QM1B with one billion training examples containing 9-11 heavy atoms. We demonstrate that a simple baseline neural network (SchNet 9M) improves its performance by simply increasing the amount of training data without additional inductive biases. To encourage future researchers to use QM1B responsibly, we highlight several limitations of QM1B and emphasise the low-resolution of our DFT options, which also serves as motivation for even larger, more accurate datasets. Code and dataset are available on Github: http://github.com/graphcore-research/pyscf-ipu
Role of Structural and Conformational Diversity for Machine Learning Potentials
Oct 30, 2023



In the field of Machine Learning Interatomic Potentials (MLIPs), understanding the intricate relationship between data biases, specifically conformational and structural diversity, and model generalization is critical in improving the quality of Quantum Mechanics (QM) data generation efforts. We investigate these dynamics through two distinct experiments: a fixed budget one, where the dataset size remains constant, and a fixed molecular set one, which focuses on fixed structural diversity while varying conformational diversity. Our results reveal nuanced patterns in generalization metrics. Notably, for optimal structural and conformational generalization, a careful balance between structural and conformational diversity is required, but existing QM datasets do not meet that trade-off. Additionally, our results highlight the limitation of the MLIP models at generalizing beyond their training distribution, emphasizing the importance of defining applicability domain during model deployment. These findings provide valuable insights and guidelines for QM data generation efforts.
Towards Foundational Models for Molecular Learning on Large-Scale Multi-Task Datasets
Oct 18, 2023



Recently, pre-trained foundation models have enabled significant advancements in multiple fields. In molecular machine learning, however, where datasets are often hand-curated, and hence typically small, the lack of datasets with labeled features, and codebases to manage those datasets, has hindered the development of foundation models. In this work, we present seven novel datasets categorized by size into three distinct categories: ToyMix, LargeMix and UltraLarge. These datasets push the boundaries in both the scale and the diversity of supervised labels for molecular learning. They cover nearly 100 million molecules and over 3000 sparsely defined tasks, totaling more than 13 billion individual labels of both quantum and biological nature. In comparison, our datasets contain 300 times more data points than the widely used OGB-LSC PCQM4Mv2 dataset, and 13 times more than the quantum-only QM1B dataset. In addition, to support the development of foundational models based on our proposed datasets, we present the Graphium graph machine learning library which simplifies the process of building and training molecular machine learning models for multi-task and multi-level molecular datasets. Finally, we present a range of baseline results as a starting point of multi-task and multi-level training on these datasets. Empirically, we observe that performance on low-resource biological datasets show improvement by also training on large amounts of quantum data. This indicates that there may be potential in multi-task and multi-level training of a foundation model and fine-tuning it to resource-constrained downstream tasks.