 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGenerating QM1B with PySCF$_{\text{IPU}}$
Nov 02, 2023The emergence of foundation models in Computer Vision and Natural Language Processing have resulted in immense progress on downstream tasks. This progress was enabled by datasets with billions of training examples. Similar benefits are yet to be unlocked for quantum chemistry, where the potential of deep learning is constrained by comparatively small datasets with 100k to 20M training examples. These datasets are limited in size because the labels are computed using the accurate (but computationally demanding) predictions of Density Functional Theory (DFT). Notably, prior DFT datasets were created using CPU supercomputers without leveraging hardware acceleration. In this paper, we take a first step towards utilising hardware accelerators by introducing the data generator PySCF$_{\text{IPU}}$ using Intelligence Processing Units (IPUs). This allowed us to create the dataset QM1B with one billion training examples containing 9-11 heavy atoms. We demonstrate that a simple baseline neural network (SchNet 9M) improves its performance by simply increasing the amount of training data without additional inductive biases. To encourage future researchers to use QM1B responsibly, we highlight several limitations of QM1B and emphasise the low-resolution of our DFT options, which also serves as motivation for even larger, more accurate datasets. Code and dataset are available on Github: http://github.com/graphcore-research/pyscf-ipu
Towards Foundational Models for Molecular Learning on Large-Scale Multi-Task Datasets
Oct 18, 2023



Recently, pre-trained foundation models have enabled significant advancements in multiple fields. In molecular machine learning, however, where datasets are often hand-curated, and hence typically small, the lack of datasets with labeled features, and codebases to manage those datasets, has hindered the development of foundation models. In this work, we present seven novel datasets categorized by size into three distinct categories: ToyMix, LargeMix and UltraLarge. These datasets push the boundaries in both the scale and the diversity of supervised labels for molecular learning. They cover nearly 100 million molecules and over 3000 sparsely defined tasks, totaling more than 13 billion individual labels of both quantum and biological nature. In comparison, our datasets contain 300 times more data points than the widely used OGB-LSC PCQM4Mv2 dataset, and 13 times more than the quantum-only QM1B dataset. In addition, to support the development of foundational models based on our proposed datasets, we present the Graphium graph machine learning library which simplifies the process of building and training molecular machine learning models for multi-task and multi-level molecular datasets. Finally, we present a range of baseline results as a starting point of multi-task and multi-level training on these datasets. Empirically, we observe that performance on low-resource biological datasets show improvement by also training on large amounts of quantum data. This indicates that there may be potential in multi-task and multi-level training of a foundation model and fine-tuning it to resource-constrained downstream tasks.
GPS++: Reviving the Art of Message Passing for Molecular Property Prediction
Feb 06, 2023



We present GPS++, a hybrid Message Passing Neural Network / Graph Transformer model for molecular property prediction. Our model integrates a well-tuned local message passing component and biased global attention with other key ideas from prior literature to achieve state-of-the-art results on large-scale molecular dataset PCQM4Mv2. Through a thorough ablation study we highlight the impact of individual components and, contrary to expectations set by recent trends, find that nearly all of the model's performance can be maintained without any use of global self-attention. We also show that our approach is significantly more accurate than prior art when 3D positional information is not available.
GPS++: An Optimised Hybrid MPNN/Transformer for Molecular Property Prediction
Dec 06, 2022



This technical report presents GPS++, the first-place solution to the Open Graph Benchmark Large-Scale Challenge (OGB-LSC 2022) for the PCQM4Mv2 molecular property prediction task. Our approach implements several key principles from the prior literature. At its core our GPS++ method is a hybrid MPNN/Transformer model that incorporates 3D atom positions and an auxiliary denoising task. The effectiveness of GPS++ is demonstrated by achieving 0.0719 mean absolute error on the independent test-challenge PCQM4Mv2 split. Thanks to Graphcore IPU acceleration, GPS++ scales to deep architectures (16 layers), training at 3 minutes per epoch, and large ensemble (112 models), completing the final predictions in 1 hour 32 minutes, well under the 4 hour inference budget allocated. Our implementation is publicly available at: https://github.com/graphcore/ogb-lsc-pcqm4mv2.
Acquisition-invariant brain MRI segmentation with informative uncertainties
Nov 07, 2021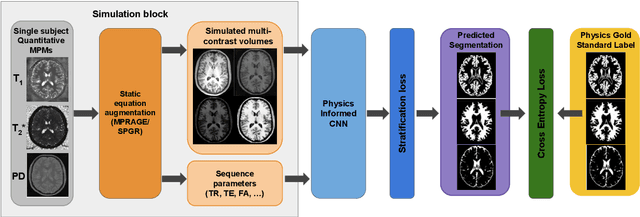
Combining multi-site data can strengthen and uncover trends, but is a task that is marred by the influence of site-specific covariates that can bias the data and therefore any downstream analyses. Post-hoc multi-site correction methods exist but have strong assumptions that often do not hold in real-world scenarios. Algorithms should be designed in a way that can account for site-specific effects, such as those that arise from sequence parameter choices, and in instances where generalisation fails, should be able to identify such a failure by means of explicit uncertainty modelling. This body of work showcases such an algorithm, that can become robust to the physics of acquisition in the context of segmentation tasks, while simultaneously modelling uncertainty. We demonstrate that our method not only generalises to complete holdout datasets, preserving segmentation quality, but does so while also accounting for site-specific sequence choices, which also allows it to perform as a harmonisation tool.
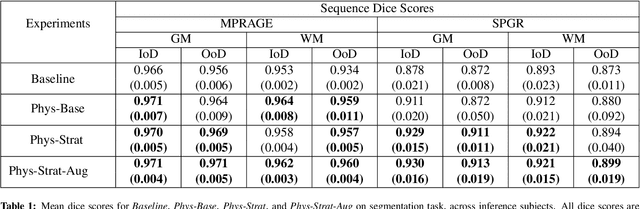
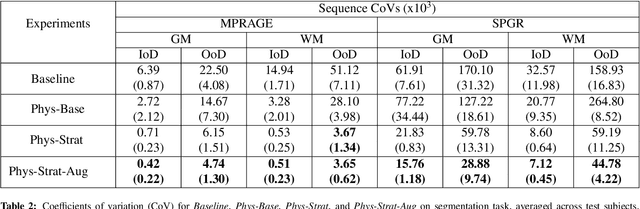
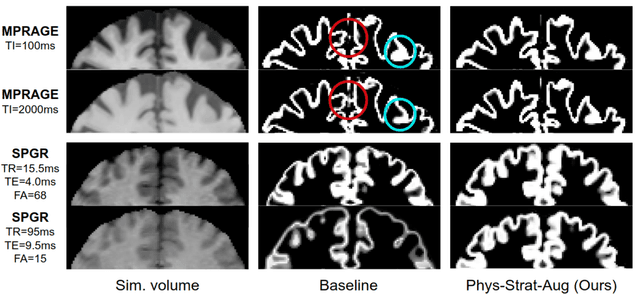
The role of MRI physics in brain segmentation CNNs: achieving acquisition invariance and instructive uncertainties
Nov 04, 2021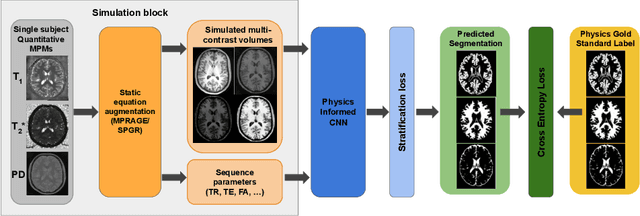
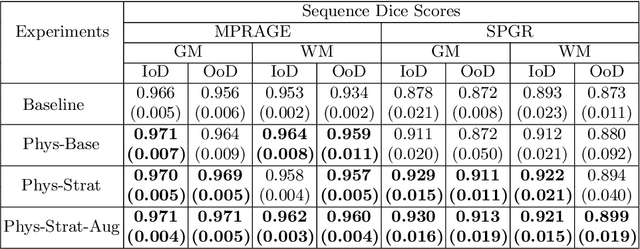
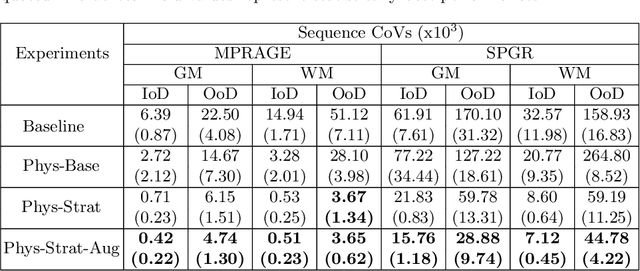
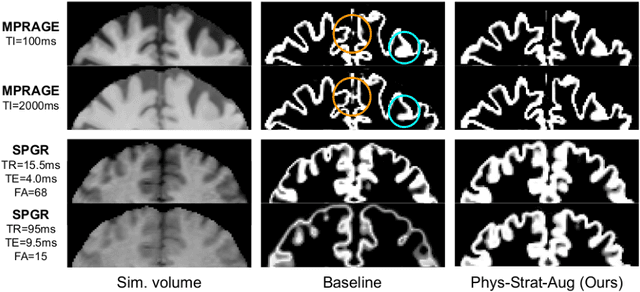
Being able to adequately process and combine data arising from different sites is crucial in neuroimaging, but is difficult, owing to site, sequence and acquisition-parameter dependent biases. It is important therefore to design algorithms that are not only robust to images of differing contrasts, but also be able to generalise well to unseen ones, with a quantifiable measure of uncertainty. In this paper we demonstrate the efficacy of a physics-informed, uncertainty-aware, segmentation network that employs augmentation-time MR simulations and homogeneous batch feature stratification to achieve acquisition invariance. We show that the proposed approach also accurately extrapolates to out-of-distribution sequence samples, providing well calibrated volumetric bounds on these. We demonstrate a significant improvement in terms of coefficients of variation, backed by uncertainty based volumetric validation.