 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCan Computational Reducibility Lead to Transferable Models for Graph Combinatorial Optimization?
Mar 02, 2026A key challenge in deriving unified neural solvers for combinatorial optimization (CO) is efficient generalization of models between a given set of tasks to new tasks not used during the initial training process. To address it, we first establish a new model, which uses a GCON module as a form of expressive message passing together with energy-based unsupervised loss functions. This model achieves high performance (often comparable with state-of-the-art results) across multiple CO tasks when trained individually on each task. We then leverage knowledge from the computational reducibility literature to propose pretraining and fine-tuning strategies that transfer effectively (a) between MVC, MIS and MaxClique, and (b) in a multi-task learning setting that additionally incorporates MaxCut, MDS and graph coloring. Additionally, in a leave-one-out, multi-task learning setting, we observe that pretraining on all but one task almost always leads to faster convergence on the remaining task when fine-tuning while avoiding negative transfer. Our findings indicate that learning common representations across multiple graph CO problems is viable through the use of expressive message passing coupled with pretraining strategies that are informed by the polynomial reduction literature, thereby taking an important step towards enabling the development of foundational models for neural CO. We provide an open-source implementation of our work at https://github.com/semihcanturk/COPT-MT .
TxPert: Leveraging Biochemical Relationships for Out-of-Distribution Transcriptomic Perturbation Prediction
May 20, 2025Accurately predicting cellular responses to genetic perturbations is essential for understanding disease mechanisms and designing effective therapies. Yet exhaustively exploring the space of possible perturbations (e.g., multi-gene perturbations or across tissues and cell types) is prohibitively expensive, motivating methods that can generalize to unseen conditions. In this work, we explore how knowledge graphs of gene-gene relationships can improve out-of-distribution (OOD) prediction across three challenging settings: unseen single perturbations; unseen double perturbations; and unseen cell lines. In particular, we present: (i) TxPert, a new state-of-the-art method that leverages multiple biological knowledge networks to predict transcriptional responses under OOD scenarios; (ii) an in-depth analysis demonstrating the impact of graphs, model architecture, and data on performance; and (iii) an expanded benchmarking framework that strengthens evaluation standards for perturbation modeling.
Towards a General GNN Framework for Combinatorial Optimization
May 31, 2024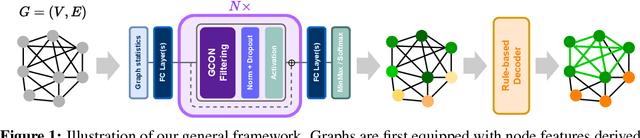
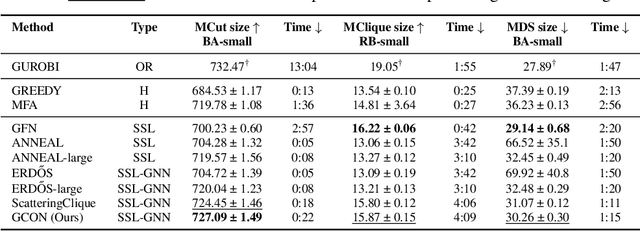
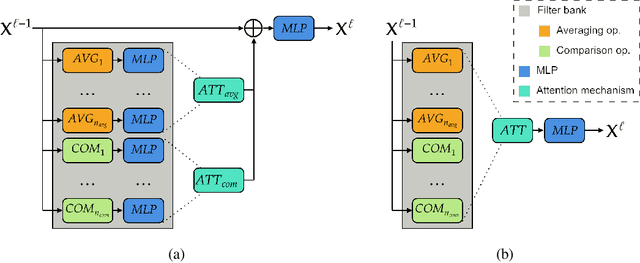
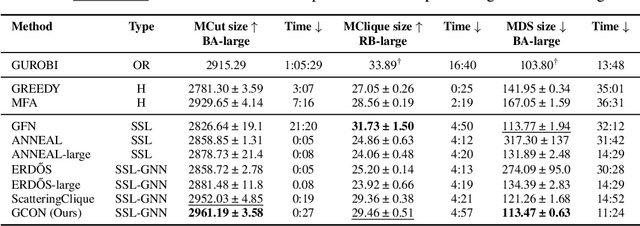
Graph neural networks (GNNs) have achieved great success for a variety of tasks such as node classification, graph classification, and link prediction. However, the use of GNNs (and machine learning more generally) to solve combinatorial optimization (CO) problems is much less explored. Here, we introduce a novel GNN architecture which leverages a complex filter bank and localized attention mechanisms designed to solve CO problems on graphs. We show how our method differentiates itself from prior GNN-based CO solvers and how it can be effectively applied to the maximum clique, minimum dominating set, and maximum cut problems in a self-supervised learning setting. In addition to demonstrating competitive overall performance across all tasks, we establish state-of-the-art results for the max cut problem.
On the Scalability of GNNs for Molecular Graphs
Apr 17, 2024

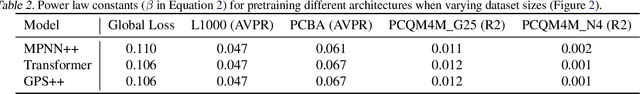
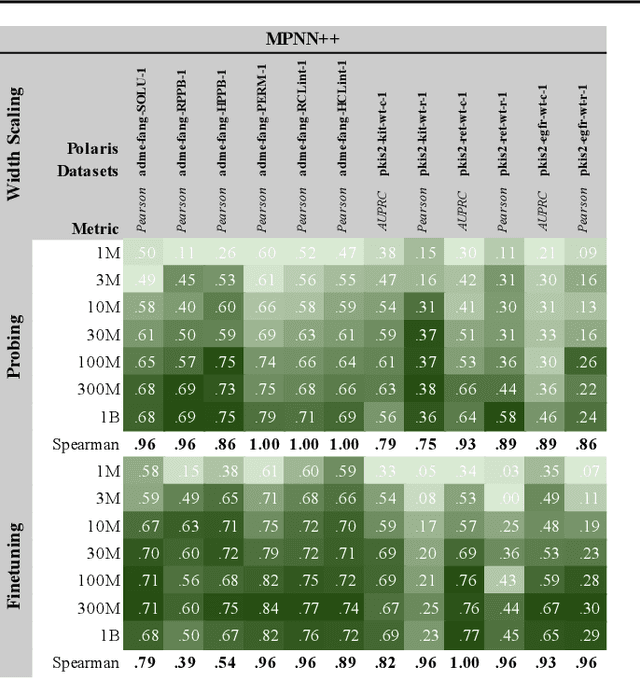
Scaling deep learning models has been at the heart of recent revolutions in language modelling and image generation. Practitioners have observed a strong relationship between model size, dataset size, and performance. However, structure-based architectures such as Graph Neural Networks (GNNs) are yet to show the benefits of scale mainly due to the lower efficiency of sparse operations, large data requirements, and lack of clarity about the effectiveness of various architectures. We address this drawback of GNNs by studying their scaling behavior. Specifically, we analyze message-passing networks, graph Transformers, and hybrid architectures on the largest public collection of 2D molecular graphs. For the first time, we observe that GNNs benefit tremendously from the increasing scale of depth, width, number of molecules, number of labels, and the diversity in the pretraining datasets, resulting in a 30.25% improvement when scaling to 1 billion parameters and 28.98% improvement when increasing size of dataset to eightfold. We further demonstrate strong finetuning scaling behavior on 38 tasks, outclassing previous large models. We hope that our work paves the way for an era where foundational GNNs drive pharmaceutical drug discovery.
Towards Foundational Models for Molecular Learning on Large-Scale Multi-Task Datasets
Oct 18, 2023



Recently, pre-trained foundation models have enabled significant advancements in multiple fields. In molecular machine learning, however, where datasets are often hand-curated, and hence typically small, the lack of datasets with labeled features, and codebases to manage those datasets, has hindered the development of foundation models. In this work, we present seven novel datasets categorized by size into three distinct categories: ToyMix, LargeMix and UltraLarge. These datasets push the boundaries in both the scale and the diversity of supervised labels for molecular learning. They cover nearly 100 million molecules and over 3000 sparsely defined tasks, totaling more than 13 billion individual labels of both quantum and biological nature. In comparison, our datasets contain 300 times more data points than the widely used OGB-LSC PCQM4Mv2 dataset, and 13 times more than the quantum-only QM1B dataset. In addition, to support the development of foundational models based on our proposed datasets, we present the Graphium graph machine learning library which simplifies the process of building and training molecular machine learning models for multi-task and multi-level molecular datasets. Finally, we present a range of baseline results as a starting point of multi-task and multi-level training on these datasets. Empirically, we observe that performance on low-resource biological datasets show improvement by also training on large amounts of quantum data. This indicates that there may be potential in multi-task and multi-level training of a foundation model and fine-tuning it to resource-constrained downstream tasks.
Inferring dynamic regulatory interaction graphs from time series data with perturbations
Jun 13, 2023


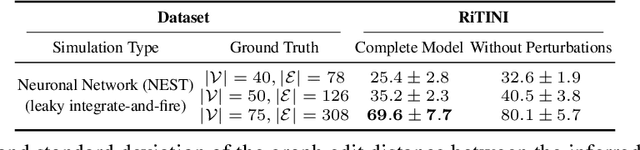
Complex systems are characterized by intricate interactions between entities that evolve dynamically over time. Accurate inference of these dynamic relationships is crucial for understanding and predicting system behavior. In this paper, we propose Regulatory Temporal Interaction Network Inference (RiTINI) for inferring time-varying interaction graphs in complex systems using a novel combination of space-and-time graph attentions and graph neural ordinary differential equations (ODEs). RiTINI leverages time-lapse signals on a graph prior, as well as perturbations of signals at various nodes in order to effectively capture the dynamics of the underlying system. This approach is distinct from traditional causal inference networks, which are limited to inferring acyclic and static graphs. In contrast, RiTINI can infer cyclic, directed, and time-varying graphs, providing a more comprehensive and accurate representation of complex systems. The graph attention mechanism in RiTINI allows the model to adaptively focus on the most relevant interactions in time and space, while the graph neural ODEs enable continuous-time modeling of the system's dynamics. We evaluate RiTINI's performance on various simulated and real-world datasets, demonstrating its state-of-the-art capability in inferring interaction graphs compared to previous methods.
Learnable Filters for Geometric Scattering Modules
Aug 15, 2022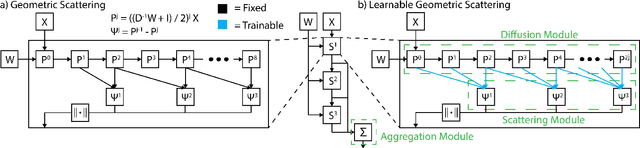
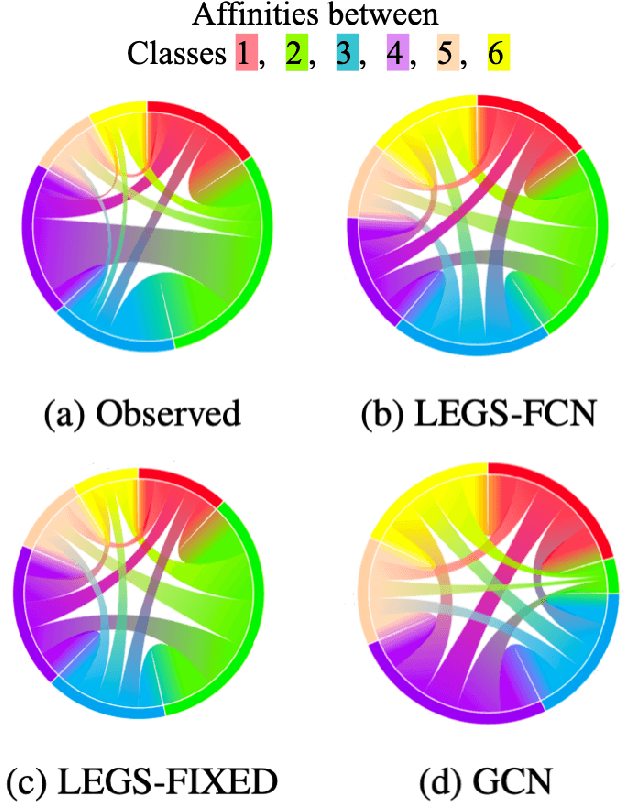
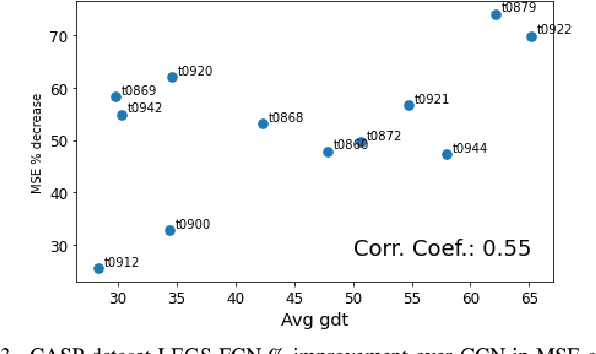
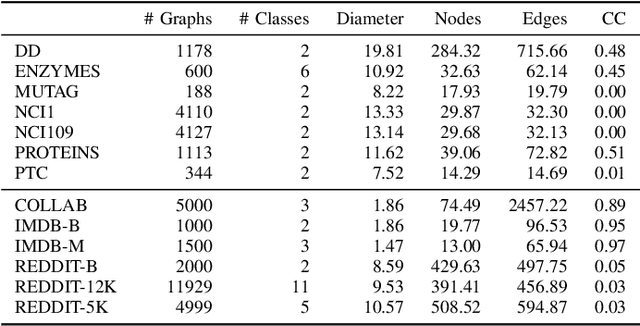
We propose a new graph neural network (GNN) module, based on relaxations of recently proposed geometric scattering transforms, which consist of a cascade of graph wavelet filters. Our learnable geometric scattering (LEGS) module enables adaptive tuning of the wavelets to encourage band-pass features to emerge in learned representations. The incorporation of our LEGS-module in GNNs enables the learning of longer-range graph relations compared to many popular GNNs, which often rely on encoding graph structure via smoothness or similarity between neighbors. Further, its wavelet priors result in simplified architectures with significantly fewer learned parameters compared to competing GNNs. We demonstrate the predictive performance of LEGS-based networks on graph classification benchmarks, as well as the descriptive quality of their learned features in biochemical graph data exploration tasks. Our results show that LEGS-based networks match or outperforms popular GNNs, as well as the original geometric scattering construction, on many datasets, in particular in biochemical domains, while retaining certain mathematical properties of handcrafted (non-learned) geometric scattering.
Taxonomy of Benchmarks in Graph Representation Learning
Jun 15, 2022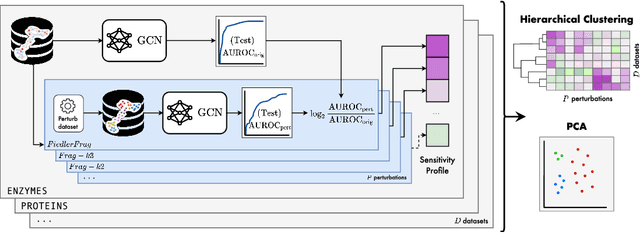
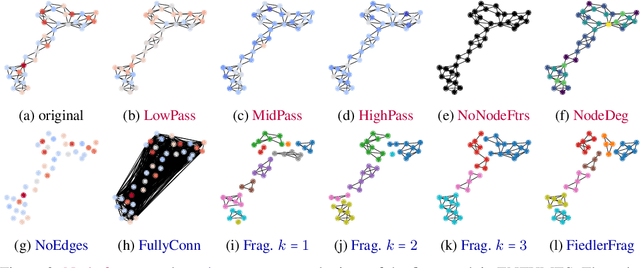
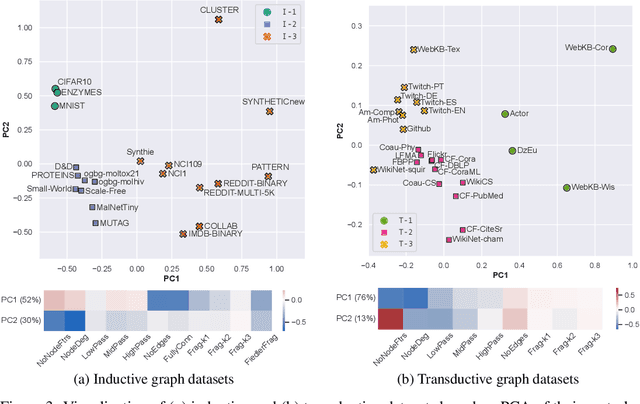
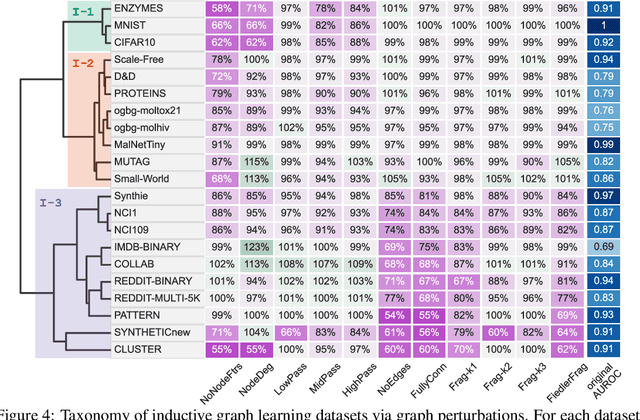
Graph Neural Networks (GNNs) extend the success of neural networks to graph-structured data by accounting for their intrinsic geometry. While extensive research has been done on developing GNN models with superior performance according to a collection of graph representation learning benchmarks, it is currently not well understood what aspects of a given model are probed by them. For example, to what extent do they test the ability of a model to leverage graph structure vs. node features? Here, we develop a principled approach to taxonomize benchmarking datasets according to a $\textit{sensitivity profile}$ that is based on how much GNN performance changes due to a collection of graph perturbations. Our data-driven analysis provides a deeper understanding of which benchmarking data characteristics are leveraged by GNNs. Consequently, our taxonomy can aid in selection and development of adequate graph benchmarks, and better informed evaluation of future GNN methods. Finally, our approach and implementation in $\texttt{GTaxoGym}$ package are extendable to multiple graph prediction task types and future datasets.
Can Hybrid Geometric Scattering Networks Help Solve the Maximal Clique Problem?
Jun 03, 2022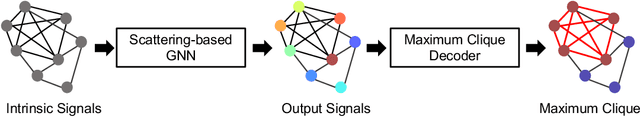
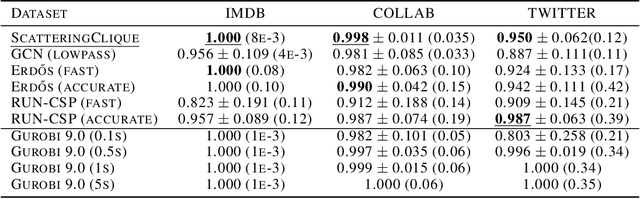
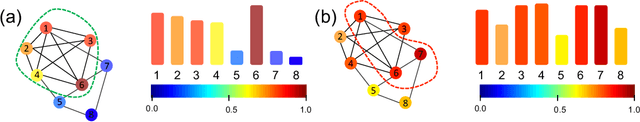
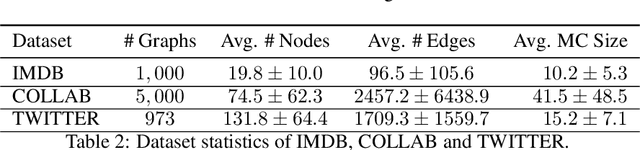
We propose a geometric scattering-based graph neural network (GNN) for approximating solutions of the NP-hard maximal clique (MC) problem. We construct a loss function with two terms, one which encourages the network to find a large set of nodes and the other which acts as a surrogate for the constraint that the nodes form a clique. We then use this loss to train a novel GNN architecture that outputs a vector representing the probability for each node to be part of the MC and apply a rule-based decoder to make our final prediction. The incorporation of the scattering transform alleviates the so-called oversmoothing problem that is often encountered in GNNs and would degrade the performance of our proposed setup. Our empirical results demonstrate that our method outperforms representative GNN baselines in terms of solution accuracy and inference speed as well as conventional solvers like GUROBI with limited time budgets.
Overcoming Oversmoothness in Graph Convolutional Networks via Hybrid Scattering Networks
Jan 22, 2022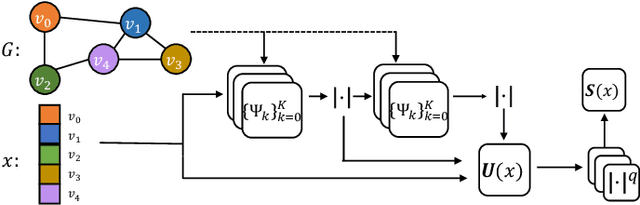
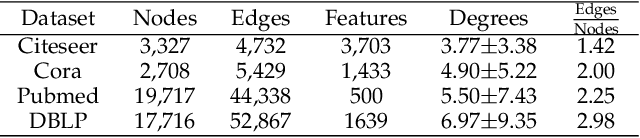
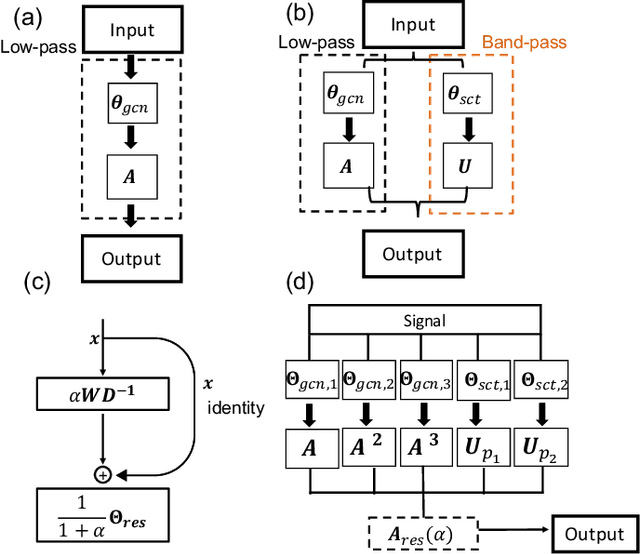
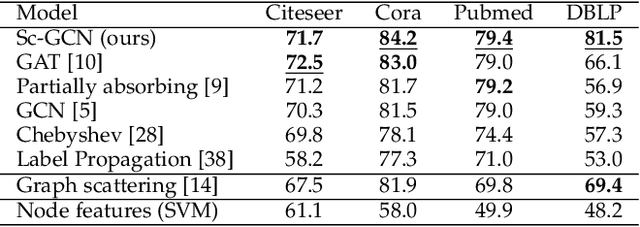
Geometric deep learning (GDL) has made great strides towards generalizing the design of structure-aware neural network architectures from traditional domains to non-Euclidean ones, such as graphs. This gave rise to graph neural network (GNN) models that can be applied to graph-structured datasets arising, for example, in social networks, biochemistry, and material science. Graph convolutional networks (GCNs) in particular, inspired by their Euclidean counterparts, have been successful in processing graph data by extracting structure-aware features. However, current GNN models (and GCNs in particular) are known to be constrained by various phenomena that limit their expressive power and ability to generalize to more complex graph datasets. Most models essentially rely on low-pass filtering of graph signals via local averaging operations, thus leading to oversmoothing. Here, we propose a hybrid GNN framework that combines traditional GCN filters with band-pass filters defined via the geometric scattering transform. We further introduce an attention framework that allows the model to locally attend over the combined information from different GNN filters at the node level. Our theoretical results establish the complementary benefits of the scattering filters to leverage structural information from the graph, while our experiments show the benefits of our method on various learning tasks.