 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAutoregressive Boltzmann Generators
Jun 25, 2026Efficient sampling of molecular systems at thermodynamic equilibrium is a hallmark challenge in statistical physics. This challenge has driven the development of Boltzmann Generators (BGs), which allow rapid generation of uncorrelated equilibrium samples by combining a generative model with exact likelihoods and an importance sampling correction. However, modern BGs predominantly rely on normalizing flows (NFs), which either suffer from limited expressivity due to strict invertibility constraints (discrete time) or computationally expensive likelihoods (continuous time). In this paper, we propose Autoregressive Boltzmann Generators (ArBG) -- a novel autoregressive modelling framework -- that overcomes these limitations by departing from the flow-based BG paradigm. ArBG circumvents the topological constraints of flows and enables sequential inference-time interventions, while offering enhanced scalability by leveraging architectures effective in Large Language Models. We empirically demonstrate that ArBG leads to significant improvements over flow-based models across all benchmarks, but particularly in larger peptide systems such as the 10-residue Chignolin. Furthermore, we introduce Robin, a 132 million parameter transferable model trained with the ArBG framework which improves over the previous state-of-the-art, reducing the zero-shot energy error, E-W$_2$, on 8-residue systems by over 60$\%$. The code can be found at the following link: https://github.com/danyalrehman/autobg.
Topological Flow Matching
Jun 14, 2026Flow matching is a powerful generative modeling framework, valued for its simplicity and strong empirical performance. However, its standard formulation treats signals on structured spaces, such as fMRI data on brain graphs, as points in Euclidean space, overlooking the rich topological features of their domains. To address this, we introduce topological flow matching, a topology-aware generalization of flow matching. We interpret flow matching as a framework for solving a degenerate Schrödinger bridge problem and inject topological information by augmenting the reference process with a Laplacian-derived drift. This principled modification captures the structure of the underlying domain while preserving the desirable properties of flow matching: a stable, simulation-free objective and deterministic sample paths. As a result, our framework serves as a drop-in replacement for standard flow matching. We demonstrate its effectiveness on diverse structured datasets, including brain fMRIs, ocean currents, seismic events, and traffic flows.
Hacking Generative Perplexity: Why Unconditional Text Evaluation Needs Distributional Metrics
Jun 07, 2026Diffusion and continuous flow-based language models have emerged as the leading non-autoregressive alternatives to language modeling. Progress in both paradigms is overwhelmingly tracked by generative perplexity (gen-PPL): the per-token negative log-likelihood of samples under a frozen autoregressive (AR) scorer such as gpt2-large, typically paired with an empirical-entropy guardrail to rule out low-entropy collapse. We argue that this metric is unsound. By construction, gen-PPL measures only predictability under the scoring AR, not grammaticality or semantic coherence -- and the set of predictable but still low-quality sequences is combinatorially large. To make this concrete, we construct a suite of zero-parameter, deliberately naive samplers that achieve state-of-the-art gen-PPL on LM1B and OpenWebText at non-degenerate entropy, surpassing recently published diffusion and continuous-flow models while producing text that is incoherent by construction. We recommend evaluation suites that directly quantify the distributional divergence between generated and reference text, and use such a suite to re-benchmark recent non-autoregressive models, recovering a more faithful picture of the current state of the art.
Why Are DMD Students Lazy? Understanding the Copying Behavior in Few-Step Distillation
Jun 01, 2026Distribution Matching Distillation (DMD) compresses pretrained diffusion models into efficient few-step generators by aligning their noised distributions across all scales. In principle, such distribution-level supervision remains agnostic to specific noise-data pairings of the teacher; this provides the student the freedom to remap latent noise, a behavior consistently observed in low-dimensional settings. Surprisingly, we find that in high-dimensional settings, distilled students spontaneously reproduce the original noise-data pairings of the teacher, a phenomenon we term copying. We demonstrate that copying is neither a byproduct of adversarial objectives nor a result of teacher memorization. Instead, our evidence suggests that copying is an emergent property arising from the limited geometric freedom of the student model during high-dimensional distillation.
Strong Stochastic Flow Maps
May 31, 2026Flow and diffusion models generate high-quality samples in many modalities; however, many network evaluations are required during inference due to numerical integration of an underlying differential equation. Flow maps alleviate this problem by learning the solution map of the differential equation directly, enabling few-step sampling. Yet, current methods are restricted to approximating the solution map of ODEs. These methods can be used to learn the transition kernel of an SDE, thereby obtaining a solution map that recovers the marginal distributions of the process (weak convergence) rather than the solution path (strong convergence). We propose Strong Stochastic Flow Maps (SSFMs) as a novel framework for learning the strong solution map of additive-noise SDEs, directly generalizing deterministic flow maps to the stochastic setting. Further, a polynomial approximation to Brownian motion is introduced and shown to converge pathwise. These results enable a simulation-free training objective for the solution map of diffusion models. We demonstrate that SSFMs outperform previous stochastic flow map methods on image generation and enable few-step sampling of molecular systems.
General Multimodal Protein Design Enables DNA-Encoding of Chemistry
Apr 06, 2026Evolution is an extraordinary engine for enzymatic diversity, yet the chemistry it has explored remains a narrow slice of what DNA can encode. Deep generative models can design new proteins that bind ligands, but none have created enzymes without pre-specifying catalytic residues. We introduce DISCO (DIffusion for Sequence-structure CO-design), a multimodal model that co-designs protein sequence and 3D structure around arbitrary biomolecules, as well as inference-time scaling methods that optimize objectives across both modalities. Conditioned solely on reactive intermediates, DISCO designs diverse heme enzymes with novel active-site geometries. These enzymes catalyze new-to-nature carbene-transfer reactions, including alkene cyclopropanation, spirocyclopropanation, B-H, and C(sp$^3$)-H insertions, with high activities exceeding those of engineered enzymes. Random mutagenesis of a selected design further confirmed that enzyme activity can be improved through directed evolution. By providing a scalable route to evolvable enzymes, DISCO broadens the potential scope of genetically encodable transformations. Code is available at https://github.com/DISCO-design/DISCO.
MIOFlow 2.0: A unified framework for inferring cellular stochastic dynamics from single cell and spatial transcriptomics data
Mar 23, 2026Understanding cellular trajectories via time-resolved single-cell transcriptomics is vital for studying development, regeneration, and disease. A key challenge is inferring continuous trajectories from discrete snapshots. Biological complexity stems from stochastic cell fate decisions, temporal proliferation changes, and spatial environmental influences. Current methods often use deterministic interpolations treating cells in isolation, failing to capture the probabilistic branching, population shifts, and niche-dependent signaling driving real biological processes. We introduce Manifold Interpolating Optimal-Transport Flow (MIOFlow) 2.0. This framework learns biologically informed cellular trajectories by integrating manifold learning, optimal transport, and neural differential equations. It models three core processes: (1) stochasticity and branching via Neural Stochastic Differential Equations; (2) non-conservative population changes using a learned growth-rate model initialized with unbalanced optimal transport; and (3) environmental influence through a joint latent space unifying gene expression with spatial features like local cell type composition and signaling. By operating in a PHATE-distance matching autoencoder latent space, MIOFlow 2.0 ensures trajectories respect the data's intrinsic geometry. Empirical comparisons show expressive trajectory learning via neural differential equations outperforms existing generative models, including simulation-free flow matching. Validated on synthetic datasets, embryoid body differentiation, and spatially resolved axolotl brain regeneration, MIOFlow 2.0 improves trajectory accuracy and reveals hidden drivers of cellular transitions, like specific signaling niches. MIOFlow 2.0 thus bridges single-cell and spatial transcriptomics to uncover tissue-scale trajectories.
MacroGuide: Topological Guidance for Macrocycle Generation
Feb 16, 2026Macrocycles are ring-shaped molecules that offer a promising alternative to small-molecule drugs due to their enhanced selectivity and binding affinity against difficult targets. Despite their chemical value, they remain underexplored in generative modeling, likely owing to their scarcity in public datasets and the challenges of enforcing topological constraints in standard deep generative models. We introduce MacroGuide: Topological Guidance for Macrocycle Generation, a diffusion guidance mechanism that uses Persistent Homology to steer the sampling of pretrained molecular generative models toward the generation of macrocycles, in both unconditional and conditional (protein pocket) settings. At each denoising step, MacroGuide constructs a Vietoris-Rips complex from atomic positions and promotes ring formation by optimizing persistent homology features. Empirically, applying MacroGuide to pretrained diffusion models increases macrocycle generation rates from 1% to 99%, while matching or exceeding state-of-the-art performance on key quality metrics such as chemical validity, diversity, and PoseBusters checks.
FALCON: Few-step Accurate Likelihoods for Continuous Flows
Dec 10, 2025

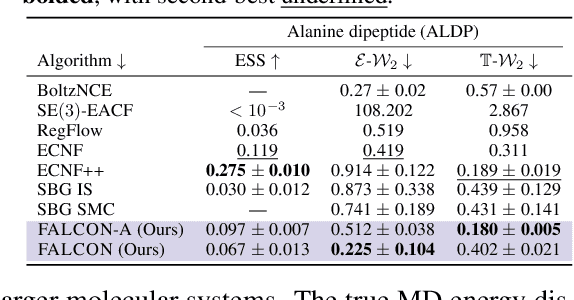

Scalable sampling of molecular states in thermodynamic equilibrium is a long-standing challenge in statistical physics. Boltzmann Generators tackle this problem by pairing a generative model, capable of exact likelihood computation, with importance sampling to obtain consistent samples under the target distribution. Current Boltzmann Generators primarily use continuous normalizing flows (CNFs) trained with flow matching for efficient training of powerful models. However, likelihood calculation for these models is extremely costly, requiring thousands of function evaluations per sample, severely limiting their adoption. In this work, we propose Few-step Accurate Likelihoods for Continuous Flows (FALCON), a method which allows for few-step sampling with a likelihood accurate enough for importance sampling applications by introducing a hybrid training objective that encourages invertibility. We show FALCON outperforms state-of-the-art normalizing flow models for molecular Boltzmann sampling and is two orders of magnitude faster than the equivalently performing CNF model.
Curly Flow Matching for Learning Non-gradient Field Dynamics
Oct 30, 2025Modeling the transport dynamics of natural processes from population-level observations is a ubiquitous problem in the natural sciences. Such models rely on key assumptions about the underlying process in order to enable faithful learning of governing dynamics that mimic the actual system behavior. The de facto assumption in current approaches relies on the principle of least action that results in gradient field dynamics and leads to trajectories minimizing an energy functional between two probability measures. However, many real-world systems, such as cell cycles in single-cell RNA, are known to exhibit non-gradient, periodic behavior, which fundamentally cannot be captured by current state-of-the-art methods such as flow and bridge matching. In this paper, we introduce Curly Flow Matching (Curly-FM), a novel approach that is capable of learning non-gradient field dynamics by designing and solving a Schr\"odinger bridge problem with a non-zero drift reference process -- in stark contrast to typical zero-drift reference processes -- which is constructed using inferred velocities in addition to population snapshot data. We showcase Curly-FM by solving the trajectory inference problems for single cells, computational fluid dynamics, and ocean currents with approximate velocities. We demonstrate that Curly-FM can learn trajectories that better match both the reference process and population marginals. Curly-FM expands flow matching models beyond the modeling of populations and towards the modeling of known periodic behavior in physical systems. Our code repository is accessible at: https://github.com/kpetrovicc/curly-flow-matching.git