 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDe novo molecular structure elucidation from mass spectra via flow matching
Feb 23, 2026Mass spectrometry is a powerful and widely used tool for identifying molecular structures due to its sensitivity and ability to profile complex samples. However, translating spectra into full molecular structures is a difficult, under-defined inverse problem. Overcoming this problem is crucial for enabling biological insight, discovering new metabolites, and advancing chemical research across multiple fields. To this end, we develop MSFlow, a two-stage encoder-decoder flow-matching generative model that achieves state-of-the-art performance on the structure elucidation task for small molecules. In the first stage, we adopt a formula-restricted transformer model for encoding mass spectra into a continuous and chemically informative embedding space, while in the second stage, we train a decoder flow matching model to reconstruct molecules from latent embeddings of mass spectra. We present ablation studies demonstrating the importance of using information-preserving molecular descriptors for encoding mass spectra and motivate the use of our discrete flow-based decoder. Our rigorous evaluation demonstrates that MSFlow can accurately translate up to 45 percent of molecular mass spectra into their corresponding molecular representations - an improvement of up to fourteen-fold over the current state-of-the-art. A trained version of MSFlow is made publicly available on GitHub for non-commercial users.
cp_measure: API-first feature extraction for image-based profiling workflows
Jul 01, 2025Biological image analysis has traditionally focused on measuring specific visual properties of interest for cells or other entities. A complementary paradigm gaining increasing traction is image-based profiling - quantifying many distinct visual features to form comprehensive profiles which may reveal hidden patterns in cellular states, drug responses, and disease mechanisms. While current tools like CellProfiler can generate these feature sets, they pose significant barriers to automated and reproducible analyses, hindering machine learning workflows. Here we introduce cp_measure, a Python library that extracts CellProfiler's core measurement capabilities into a modular, API-first tool designed for programmatic feature extraction. We demonstrate that cp_measure features retain high fidelity with CellProfiler features while enabling seamless integration with the scientific Python ecosystem. Through applications to 3D astrocyte imaging and spatial transcriptomics, we showcase how cp_measure enables reproducible, automated image-based profiling pipelines that scale effectively for machine learning applications in computational biology.
Targeted AMP generation through controlled diffusion with efficient embeddings
Apr 24, 2025Deep learning-based antimicrobial peptide (AMP) discovery faces critical challenges such as low experimental hit rates as well as the need for nuanced controllability and efficient modeling of peptide properties. To address these challenges, we introduce OmegAMP, a framework that leverages a diffusion-based generative model with efficient low-dimensional embeddings, precise controllability mechanisms, and novel classifiers with drastically reduced false positive rates for candidate filtering. OmegAMP enables the targeted generation of AMPs with specific physicochemical properties, activity profiles, and species-specific effectiveness. Moreover, it maximizes sample diversity while ensuring faithfulness to the underlying data distribution during generation. We demonstrate that OmegAMP achieves state-of-the-art performance across all stages of the AMP discovery pipeline, significantly advancing the potential of computational frameworks in combating antimicrobial resistance.
Unified Guidance for Geometry-Conditioned Molecular Generation
Jan 05, 2025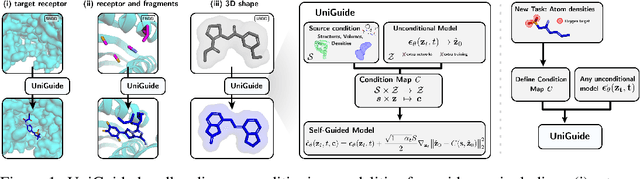
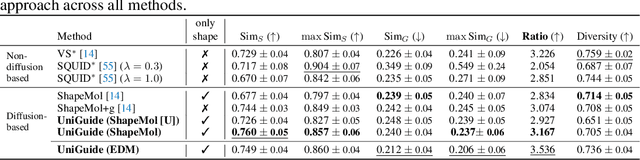
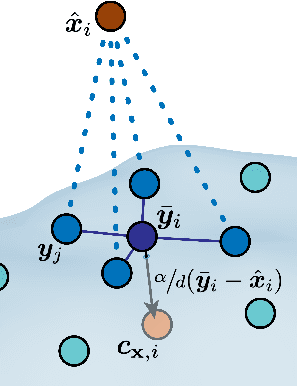
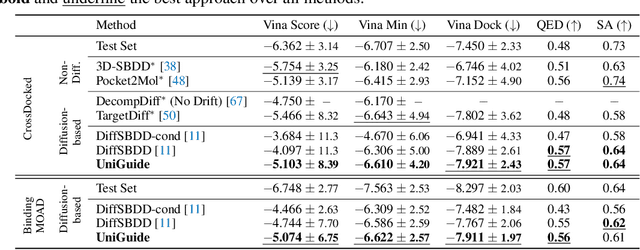
Effectively designing molecular geometries is essential to advancing pharmaceutical innovations, a domain, which has experienced great attention through the success of generative models and, in particular, diffusion models. However, current molecular diffusion models are tailored towards a specific downstream task and lack adaptability. We introduce UniGuide, a framework for controlled geometric guidance of unconditional diffusion models that allows flexible conditioning during inference without the requirement of extra training or networks. We show how applications such as structure-based, fragment-based, and ligand-based drug design are formulated in the UniGuide framework and demonstrate on-par or superior performance compared to specialised models. Offering a more versatile approach, UniGuide has the potential to streamline the development of molecular generative models, allowing them to be readily used in diverse application scenarios.
Centaur: a foundation model of human cognition
Oct 26, 2024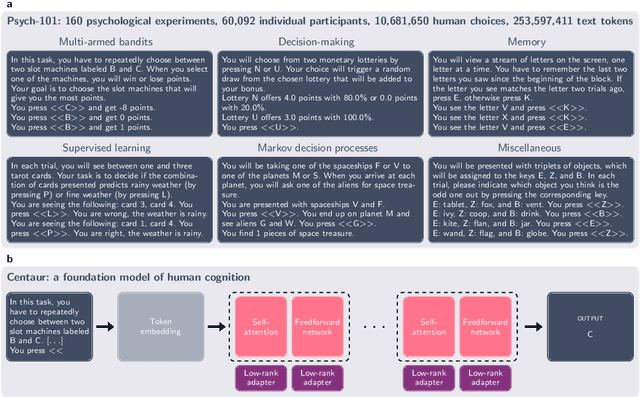

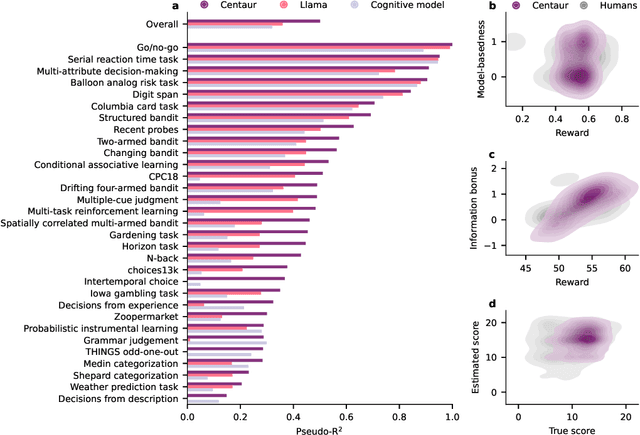
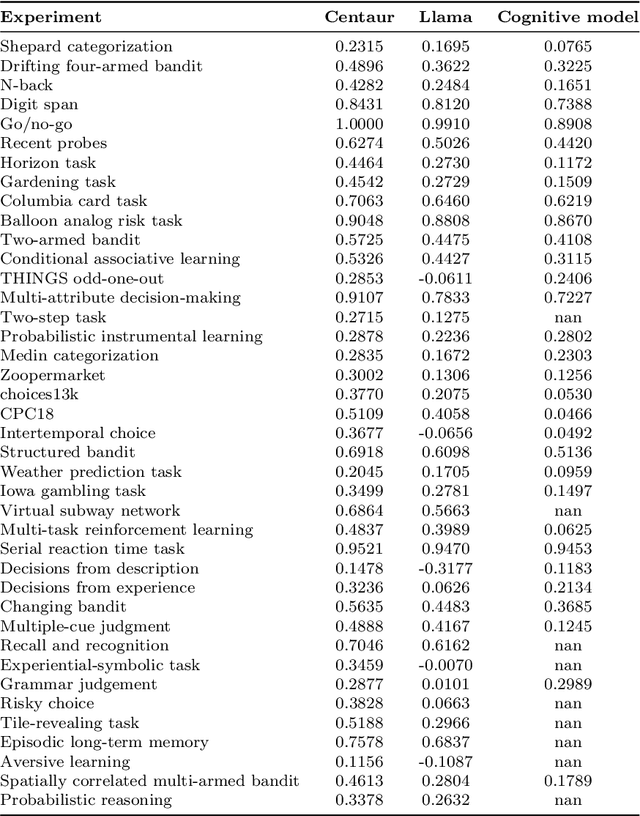
Establishing a unified theory of cognition has been a major goal of psychology. While there have been previous attempts to instantiate such theories by building computational models, we currently do not have one model that captures the human mind in its entirety. Here we introduce Centaur, a computational model that can predict and simulate human behavior in any experiment expressible in natural language. We derived Centaur by finetuning a state-of-the-art language model on a novel, large-scale data set called Psych-101. Psych-101 reaches an unprecedented scale, covering trial-by-trial data from over 60,000 participants performing over 10,000,000 choices in 160 experiments. Centaur not only captures the behavior of held-out participants better than existing cognitive models, but also generalizes to new cover stories, structural task modifications, and entirely new domains. Furthermore, we find that the model's internal representations become more aligned with human neural activity after finetuning. Taken together, Centaur is the first real candidate for a unified model of human cognition. We anticipate that it will have a disruptive impact on the cognitive sciences, challenging the existing paradigm for developing computational models.
How to Build the Virtual Cell with Artificial Intelligence: Priorities and Opportunities
Sep 18, 2024

The cell is arguably the smallest unit of life and is central to understanding biology. Accurate modeling of cells is important for this understanding as well as for determining the root causes of disease. Recent advances in artificial intelligence (AI), combined with the ability to generate large-scale experimental data, present novel opportunities to model cells. Here we propose a vision of AI-powered Virtual Cells, where robust representations of cells and cellular systems under different conditions are directly learned from growing biological data across measurements and scales. We discuss desired capabilities of AI Virtual Cells, including generating universal representations of biological entities across scales, and facilitating interpretable in silico experiments to predict and understand their behavior using Virtual Instruments. We further address the challenges, opportunities and requirements to realize this vision including data needs, evaluation strategies, and community standards and engagement to ensure biological accuracy and broad utility. We envision a future where AI Virtual Cells help identify new drug targets, predict cellular responses to perturbations, as well as scale hypothesis exploration. With open science collaborations across the biomedical ecosystem that includes academia, philanthropy, and the biopharma and AI industries, a comprehensive predictive understanding of cell mechanisms and interactions is within reach.
Generating Multi-Modal and Multi-Attribute Single-Cell Counts with CFGen
Jul 16, 2024Generative modeling of single-cell RNA-seq data has shown invaluable potential in community-driven tasks such as trajectory inference, batch effect removal and gene expression generation. However, most recent deep models generating synthetic single cells from noise operate on pre-processed continuous gene expression approximations, ignoring the inherently discrete and over-dispersed nature of single-cell data, which limits downstream applications and hinders the incorporation of robust noise models. Moreover, crucial aspects of deep-learning-based synthetic single-cell generation remain underexplored, such as controllable multi-modal and multi-label generation and its role in the performance enhancement of downstream tasks. This work presents Cell Flow for Generation (CFGen), a flow-based conditional generative model for multi-modal single-cell counts, which explicitly accounts for the discrete nature of the data. Our results suggest improved recovery of crucial biological data characteristics while accounting for novel generative tasks such as conditioning on multiple attributes and boosting rare cell type classification via data augmentation. By showcasing CFGen on a diverse set of biological datasets and settings, we provide evidence of its value to the fields of computational biology and deep generative models.
Disentangled Representation Learning through Geometry Preservation with the Gromov-Monge Gap
Jul 10, 2024



Learning disentangled representations in an unsupervised manner is a fundamental challenge in machine learning. Solving it may unlock other problems, such as generalization, interpretability, or fairness. While remarkably difficult to solve in general, recent works have shown that disentanglement is provably achievable under additional assumptions that can leverage geometrical constraints, such as local isometry. To use these insights, we propose a novel perspective on disentangled representation learning built on quadratic optimal transport. Specifically, we formulate the problem in the Gromov-Monge setting, which seeks isometric mappings between distributions supported on different spaces. We propose the Gromov-Monge-Gap (GMG), a regularizer that quantifies the geometry-preservation of an arbitrary push-forward map between two distributions supported on different spaces. We demonstrate the effectiveness of GMG regularization for disentanglement on four standard benchmarks. Moreover, we show that geometry preservation can even encourage unsupervised disentanglement without the standard reconstruction objective - making the underlying model decoder-free, and promising a more practically viable and scalable perspective on unsupervised disentanglement.
Unbalancedness in Neural Monge Maps Improves Unpaired Domain Translation
Nov 25, 2023In optimal transport (OT), a Monge map is known as a mapping that transports a source distribution to a target distribution in the most cost-efficient way. Recently, multiple neural estimators for Monge maps have been developed and applied in diverse unpaired domain translation tasks, e.g. in single-cell biology and computer vision. However, the classic OT framework enforces mass conservation, which makes it prone to outliers and limits its applicability in real-world scenarios. The latter can be particularly harmful in OT domain translation tasks, where the relative position of a sample within a distribution is explicitly taken into account. While unbalanced OT tackles this challenge in the discrete setting, its integration into neural Monge map estimators has received limited attention. We propose a theoretically grounded method to incorporate unbalancedness into any Monge map estimator. We improve existing estimators to model cell trajectories over time and to predict cellular responses to perturbations. Moreover, our approach seamlessly integrates with the OT flow matching (OT-FM) framework. While we show that OT-FM performs competitively in image translation, we further improve performance by incorporating unbalancedness (UOT-FM), which better preserves relevant features. We hence establish UOT-FM as a principled method for unpaired image translation.
Generative Entropic Neural Optimal Transport To Map Within and Across Spaces
Oct 16, 2023Learning measure-to-measure mappings is a crucial task in machine learning, featured prominently in generative modeling. Recent years have witnessed a surge of techniques that draw inspiration from optimal transport (OT) theory. Combined with neural network models, these methods collectively known as \textit{Neural OT} use optimal transport as an inductive bias: such mappings should be optimal w.r.t. a given cost function, in the sense that they are able to move points in a thrifty way, within (by minimizing displacements) or across spaces (by being isometric). This principle, while intuitive, is often confronted with several practical challenges that require adapting the OT toolbox: cost functions other than the squared-Euclidean cost can be challenging to handle, the deterministic formulation of Monge maps leaves little flexibility, mapping across incomparable spaces raises multiple challenges, while the mass conservation constraint inherent to OT can provide too much credit to outliers. While each of these mismatches between practice and theory has been addressed independently in various works, we propose in this work an elegant framework to unify them, called \textit{generative entropic neural optimal transport} (GENOT). GENOT can accommodate any cost function; handles randomness using conditional generative models; can map points across incomparable spaces, and can be used as an \textit{unbalanced} solver. We evaluate our approach through experiments conducted on various synthetic datasets and demonstrate its practicality in single-cell biology. In this domain, GENOT proves to be valuable for tasks such as modeling cell development, predicting cellular responses to drugs, and translating between different data modalities of cells.