 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEnforcing Latent Euclidean Geometry in Single-Cell VAEs for Manifold Interpolation
Jul 15, 2025Latent space interpolations are a powerful tool for navigating deep generative models in applied settings. An example is single-cell RNA sequencing, where existing methods model cellular state transitions as latent space interpolations with variational autoencoders, often assuming linear shifts and Euclidean geometry. However, unless explicitly enforced, linear interpolations in the latent space may not correspond to geodesic paths on the data manifold, limiting methods that assume Euclidean geometry in the data representations. We introduce FlatVI, a novel training framework that regularises the latent manifold of discrete-likelihood variational autoencoders towards Euclidean geometry, specifically tailored for modelling single-cell count data. By encouraging straight lines in the latent space to approximate geodesic interpolations on the decoded single-cell manifold, FlatVI enhances compatibility with downstream approaches that assume Euclidean latent geometry. Experiments on synthetic data support the theoretical soundness of our approach, while applications to time-resolved single-cell RNA sequencing data demonstrate improved trajectory reconstruction and manifold interpolation.
Targeted AMP generation through controlled diffusion with efficient embeddings
Apr 24, 2025Deep learning-based antimicrobial peptide (AMP) discovery faces critical challenges such as low experimental hit rates as well as the need for nuanced controllability and efficient modeling of peptide properties. To address these challenges, we introduce OmegAMP, a framework that leverages a diffusion-based generative model with efficient low-dimensional embeddings, precise controllability mechanisms, and novel classifiers with drastically reduced false positive rates for candidate filtering. OmegAMP enables the targeted generation of AMPs with specific physicochemical properties, activity profiles, and species-specific effectiveness. Moreover, it maximizes sample diversity while ensuring faithfulness to the underlying data distribution during generation. We demonstrate that OmegAMP achieves state-of-the-art performance across all stages of the AMP discovery pipeline, significantly advancing the potential of computational frameworks in combating antimicrobial resistance.
Unified Guidance for Geometry-Conditioned Molecular Generation
Jan 05, 2025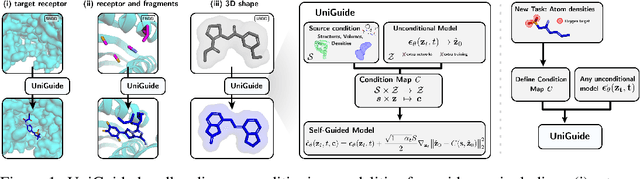
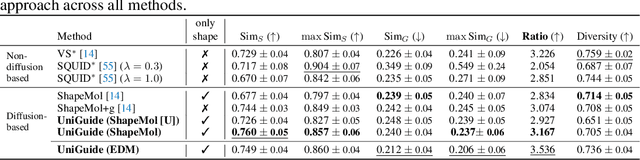
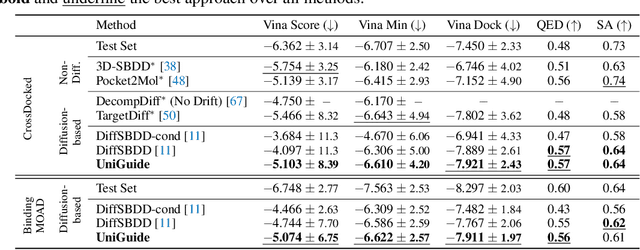
Effectively designing molecular geometries is essential to advancing pharmaceutical innovations, a domain, which has experienced great attention through the success of generative models and, in particular, diffusion models. However, current molecular diffusion models are tailored towards a specific downstream task and lack adaptability. We introduce UniGuide, a framework for controlled geometric guidance of unconditional diffusion models that allows flexible conditioning during inference without the requirement of extra training or networks. We show how applications such as structure-based, fragment-based, and ligand-based drug design are formulated in the UniGuide framework and demonstrate on-par or superior performance compared to specialised models. Offering a more versatile approach, UniGuide has the potential to streamline the development of molecular generative models, allowing them to be readily used in diverse application scenarios.
Expressivity and Generalization: Fragment-Biases for Molecular GNNs
Jun 12, 2024Although recent advances in higher-order Graph Neural Networks (GNNs) improve the theoretical expressiveness and molecular property predictive performance, they often fall short of the empirical performance of models that explicitly use fragment information as inductive bias. However, for these approaches, there exists no theoretic expressivity study. In this work, we propose the Fragment-WL test, an extension to the well-known Weisfeiler & Leman (WL) test, which enables the theoretic analysis of these fragment-biased GNNs. Building on the insights gained from the Fragment-WL test, we develop a new GNN architecture and a fragmentation with infinite vocabulary that significantly boosts expressiveness. We show the effectiveness of our model on synthetic and real-world data where we outperform all GNNs on Peptides and have 12% lower error than all GNNs on ZINC and 34% lower error than other fragment-biased models. Furthermore, we show that our model exhibits superior generalization capabilities compared to the latest transformer-based architectures, positioning it as a robust solution for a range of molecular modeling tasks.
MAGNet: Motif-Agnostic Generation of Molecules from Shapes
May 30, 2023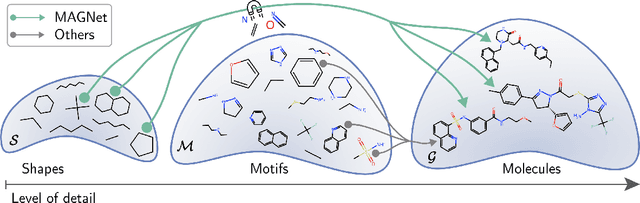
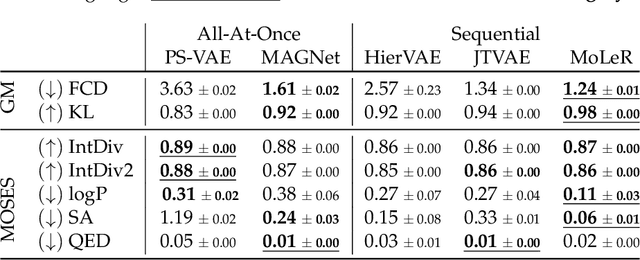
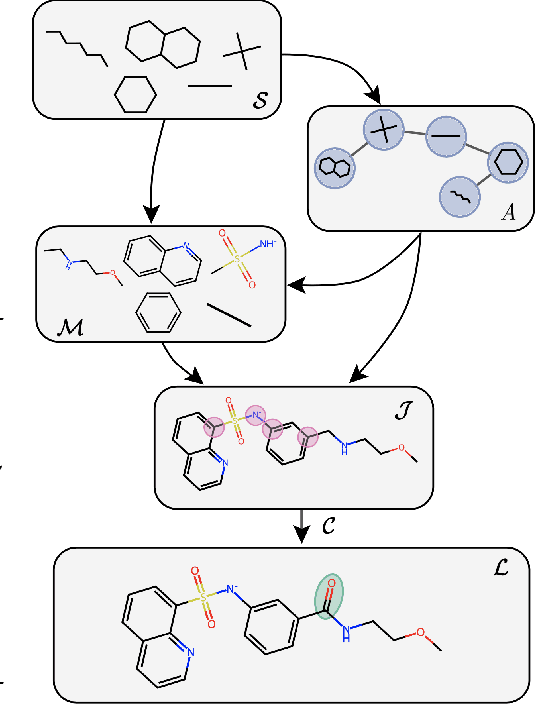
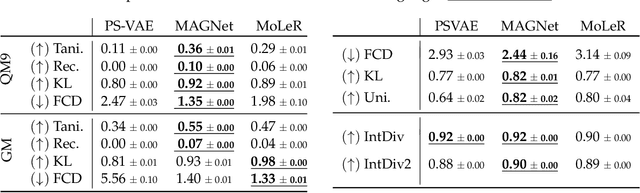
Recent advances in machine learning for molecules exhibit great potential for facilitating drug discovery from in silico predictions. Most models for molecule generation rely on the decomposition of molecules into frequently occurring substructures (motifs), from which they generate novel compounds. While motif representations greatly aid in learning molecular distributions, such methods struggle to represent substructures beyond their known motif set. To alleviate this issue and increase flexibility across datasets, we propose MAGNet, a graph-based model that generates abstract shapes before allocating atom and bond types. To this end, we introduce a novel factorisation of the molecules' data distribution that accounts for the molecules' global context and facilitates learning adequate assignments of atoms and bonds onto shapes. While the abstraction to shapes introduces greater complexity for distribution learning, we show the competitive performance of MAGNet on standard benchmarks. Importantly, we demonstrate that MAGNet's improved expressivity leads to molecules with more topologically distinct structures and, at the same time, diverse atom and bond assignments.
Uncertainty Quantification for Atlas-Level Cell Type Transfer
Nov 07, 2022



Single-cell reference atlases are large-scale, cell-level maps that capture cellular heterogeneity within an organ using single cell genomics. Given their size and cellular diversity, these atlases serve as high-quality training data for the transfer of cell type labels to new datasets. Such label transfer, however, must be robust to domain shifts in gene expression due to measurement technique, lab specifics and more general batch effects. This requires methods that provide uncertainty estimates on the cell type predictions to ensure correct interpretation. Here, for the first time, we introduce uncertainty quantification methods for cell type classification on single-cell reference atlases. We benchmark four model classes and show that currently used models lack calibration, robustness, and actionable uncertainty scores. Furthermore, we demonstrate how models that quantify uncertainty are better suited to detect unseen cell types in the setting of atlas-level cell type transfer.
Predicting single-cell perturbation responses for unseen drugs
Apr 28, 2022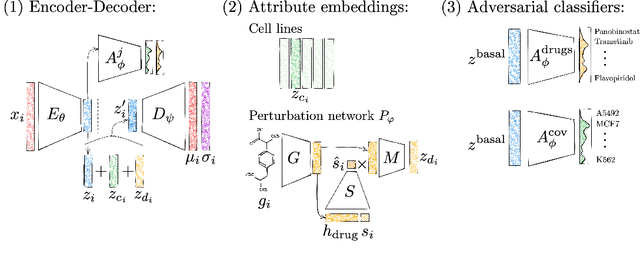
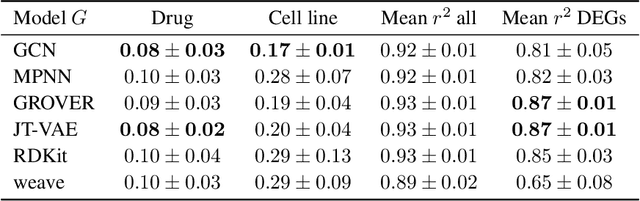
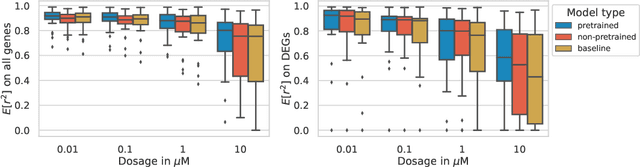
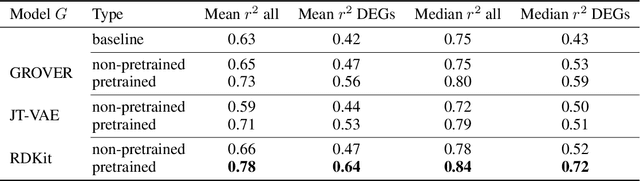
Single-cell transcriptomics enabled the study of cellular heterogeneity in response to perturbations at the resolution of individual cells. However, scaling high-throughput screens (HTSs) to measure cellular responses for many drugs remains a challenge due to technical limitations and, more importantly, the cost of such multiplexed experiments. Thus, transferring information from routinely performed bulk RNA-seq HTS is required to enrich single-cell data meaningfully. We introduce a new encoder-decoder architecture to study the perturbational effects of unseen drugs. We combine the model with a transfer learning scheme and demonstrate how training on existing bulk RNA-seq HTS datasets can improve generalisation performance. Better generalisation reduces the need for extensive and costly screens at single-cell resolution. We envision that our proposed method will facilitate more efficient experiment designs through its ability to generate in-silico hypotheses, ultimately accelerating targeted drug discovery.
* 8 pages
Interpretable and Fine-Grained Visual Explanations for Convolutional Neural Networks
Aug 07, 2019
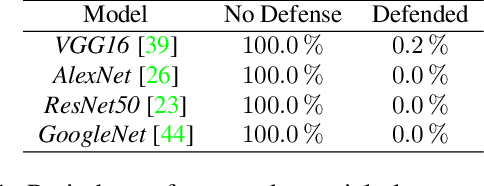

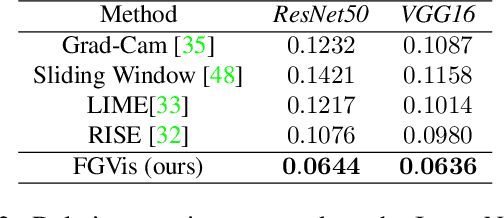
To verify and validate networks, it is essential to gain insight into their decisions, limitations as well as possible shortcomings of training data. In this work, we propose a post-hoc, optimization based visual explanation method, which highlights the evidence in the input image for a specific prediction. Our approach is based on a novel technique to defend against adversarial evidence (i.e. faulty evidence due to artefacts) by filtering gradients during optimization. The defense does not depend on human-tuned parameters. It enables explanations which are both fine-grained and preserve the characteristics of images, such as edges and colors. The explanations are interpretable, suited for visualizing detailed evidence and can be tested as they are valid model inputs. We qualitatively and quantitatively evaluate our approach on a multitude of models and datasets.