 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeChem-PerturBridge: a harmonized compendium of small molecule perturbation transcriptomic effects
May 29, 2026Large perturbation models require training data encompassing chemical, cellular, and assay diversity. Current transcriptomic resources for small-molecule modeling, however, are fragmented across technologies, metadata conventions, controls, doses, and preprocessing pipelines. We introduce Chem-PerturBridge, a harmonized multi-dataset resource comprising over 37k compounds, 136 cellular contexts, and 1.25M transcriptomic samples across eight assay types, with standardized identifiers, metadata, and replicate-aware condition-level effects. We use the resource to evaluate matched-condition agreement across datasets and replicate agreement within datasets. Matched same-compound conditions generally show weak agreement in fine-grained logFC rankings and magnitudes across most dataset pairs, often falling below same-context different-compound baselines. In contrast, logFC direction agreement is substantially more stable and usually exceeds these baselines. We further evaluate Chem-PerturBridge as a pretraining resource for compound representation learning. Under a compound-held-out OP3 evaluation split, embeddings pretrained on Chem-PerturBridge improve over L1000-only embeddings, Morgan fingerprints, and the descriptor-free OP3 baseline across metrics. An extensive molecule-holdout evaluation across 11 datasets further shows that models trained on Chem-PerturBridge outperform or match those that are not. Chem-PerturBridge therefore supports both diagnostic evaluation of cross-dataset signature agreement and model-oriented reuse of heterogeneous perturbation transcriptomic data.
annbatch unlocks terabyte-scale training of biological data in anndata
Apr 02, 2026The scale of biological datasets now routinely exceeds system memory, making data access rather than model computation the primary bottleneck in training machine-learning models. This bottleneck is particularly acute in biology, where widely used community data formats must support heterogeneous metadata, sparse and dense assays, and downstream analysis within established computational ecosystems. Here we present annbatch, a mini-batch loader native to anndata that enables out-of-core training directly on disk-backed datasets. Across single-cell transcriptomics, microscopy and whole-genome sequencing benchmarks, annbatch increases loading throughput by up to an order of magnitude and shortens training from days to hours, while remaining fully compatible with the scverse ecosystem. Annbatch establishes a practical data-loading infrastructure for scalable biological AI, allowing increasingly large and diverse datasets to be used without abandoning standard biological data formats. Github: https://github.com/scverse/annbatch
Flow-Based Density Ratio Estimation for Intractable Distributions with Applications in Genomics
Feb 27, 2026Estimating density ratios between pairs of intractable data distributions is a core problem in probabilistic modeling, enabling principled comparisons of sample likelihoods under different data-generating processes across conditions and covariates. While exact-likelihood models such as normalizing flows offer a promising approach to density ratio estimation, naive flow-based evaluations are computationally expensive, as they require simulating costly likelihood integrals for each distribution separately. In this work, we leverage condition-aware flow matching to derive a single dynamical formulation for tracking density ratios along generative trajectories. We demonstrate competitive performance on simulated benchmarks for closed-form ratio estimation, and show that our method supports versatile tasks in single-cell genomics data analysis, where likelihood-based comparisons of cellular states across experimental conditions enable treatment effect estimation and batch correction evaluation.
Enforcing Latent Euclidean Geometry in Single-Cell VAEs for Manifold Interpolation
Jul 15, 2025Latent space interpolations are a powerful tool for navigating deep generative models in applied settings. An example is single-cell RNA sequencing, where existing methods model cellular state transitions as latent space interpolations with variational autoencoders, often assuming linear shifts and Euclidean geometry. However, unless explicitly enforced, linear interpolations in the latent space may not correspond to geodesic paths on the data manifold, limiting methods that assume Euclidean geometry in the data representations. We introduce FlatVI, a novel training framework that regularises the latent manifold of discrete-likelihood variational autoencoders towards Euclidean geometry, specifically tailored for modelling single-cell count data. By encouraging straight lines in the latent space to approximate geodesic interpolations on the decoded single-cell manifold, FlatVI enhances compatibility with downstream approaches that assume Euclidean latent geometry. Experiments on synthetic data support the theoretical soundness of our approach, while applications to time-resolved single-cell RNA sequencing data demonstrate improved trajectory reconstruction and manifold interpolation.
A scalable gene network model of regulatory dynamics in single cells
Mar 25, 2025Single-cell data provide high-dimensional measurements of the transcriptional states of cells, but extracting insights into the regulatory functions of genes, particularly identifying transcriptional mechanisms affected by biological perturbations, remains a challenge. Many perturbations induce compensatory cellular responses, making it difficult to distinguish direct from indirect effects on gene regulation. Modeling how gene regulatory functions shape the temporal dynamics of these responses is key to improving our understanding of biological perturbations. Dynamical models based on differential equations offer a principled way to capture transcriptional dynamics, but their application to single-cell data has been hindered by computational constraints, stochasticity, sparsity, and noise. Existing methods either rely on low-dimensional representations or make strong simplifying assumptions, limiting their ability to model transcriptional dynamics at scale. We introduce a Functional and Learnable model of Cell dynamicS, FLeCS, that incorporates gene network structure into coupled differential equations to model gene regulatory functions. Given (pseudo)time-series single-cell data, FLeCS accurately infers cell dynamics at scale, provides improved functional insights into transcriptional mechanisms perturbed by gene knockouts, both in myeloid differentiation and K562 Perturb-seq experiments, and simulates single-cell trajectories of A549 cells following small-molecule perturbations.
B-Cos Aligned Transformers Learn Human-Interpretable Features
Jan 18, 2024Vision Transformers (ViTs) and Swin Transformers (Swin) are currently state-of-the-art in computational pathology. However, domain experts are still reluctant to use these models due to their lack of interpretability. This is not surprising, as critical decisions need to be transparent and understandable. The most common approach to understanding transformers is to visualize their attention. However, attention maps of ViTs are often fragmented, leading to unsatisfactory explanations. Here, we introduce a novel architecture called the B-cos Vision Transformer (BvT) that is designed to be more interpretable. It replaces all linear transformations with the B-cos transform to promote weight-input alignment. In a blinded study, medical experts clearly ranked BvTs above ViTs, suggesting that our network is better at capturing biomedically relevant structures. This is also true for the B-cos Swin Transformer (Bwin). Compared to the Swin Transformer, it even improves the F1-score by up to 4.7% on two public datasets.
To Transformers and Beyond: Large Language Models for the Genome
Nov 13, 2023In the rapidly evolving landscape of genomics, deep learning has emerged as a useful tool for tackling complex computational challenges. This review focuses on the transformative role of Large Language Models (LLMs), which are mostly based on the transformer architecture, in genomics. Building on the foundation of traditional convolutional neural networks and recurrent neural networks, we explore both the strengths and limitations of transformers and other LLMs for genomics. Additionally, we contemplate the future of genomic modeling beyond the transformer architecture based on current trends in research. The paper aims to serve as a guide for computational biologists and computer scientists interested in LLMs for genomic data. We hope the paper can also serve as an educational introduction and discussion for biologists to a fundamental shift in how we will be analyzing genomic data in the future.
Causal machine learning for single-cell genomics
Oct 23, 2023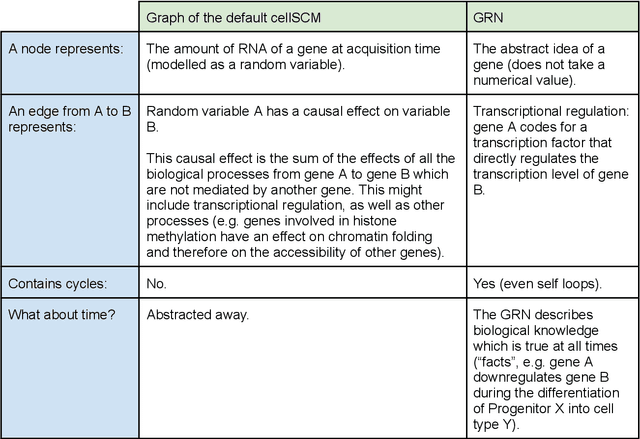
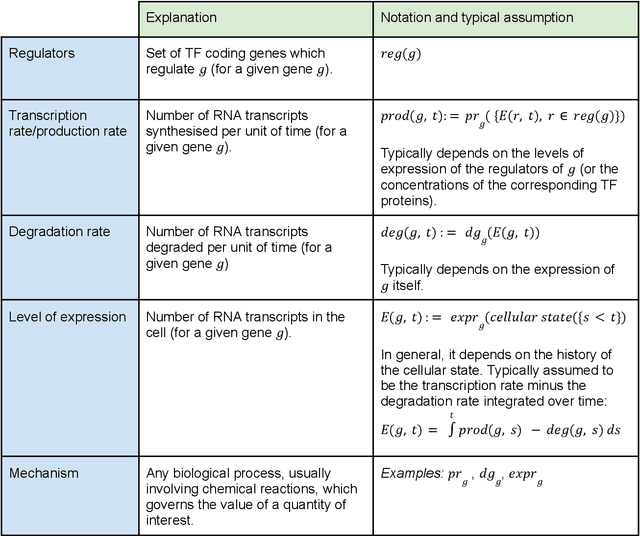
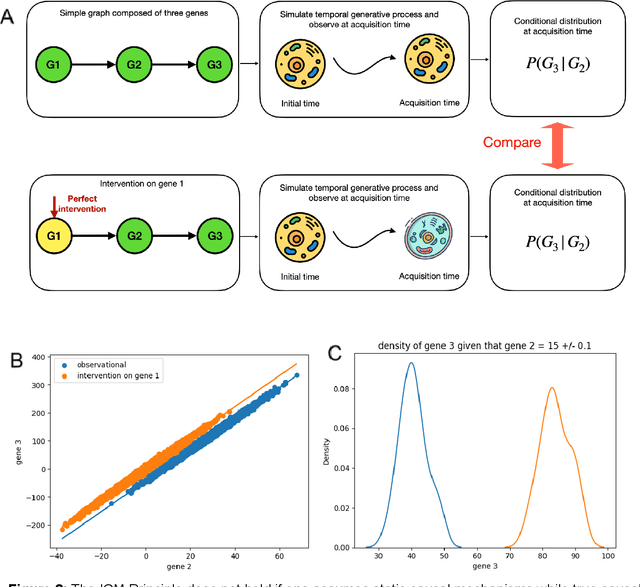

Advances in single-cell omics allow for unprecedented insights into the transcription profiles of individual cells. When combined with large-scale perturbation screens, through which specific biological mechanisms can be targeted, these technologies allow for measuring the effect of targeted perturbations on the whole transcriptome. These advances provide an opportunity to better understand the causative role of genes in complex biological processes such as gene regulation, disease progression or cellular development. However, the high-dimensional nature of the data, coupled with the intricate complexity of biological systems renders this task nontrivial. Within the machine learning community, there has been a recent increase of interest in causality, with a focus on adapting established causal techniques and algorithms to handle high-dimensional data. In this perspective, we delineate the application of these methodologies within the realm of single-cell genomics and their challenges. We first present the model that underlies most of current causal approaches to single-cell biology and discuss and challenge the assumptions it entails from the biological point of view. We then identify open problems in the application of causal approaches to single-cell data: generalising to unseen environments, learning interpretable models, and learning causal models of dynamics. For each problem, we discuss how various research directions - including the development of computational approaches and the adaptation of experimental protocols - may offer ways forward, or on the contrary pose some difficulties. With the advent of single cell atlases and increasing perturbation data, we expect causal models to become a crucial tool for informed experimental design.
Conditionally Invariant Representation Learning for Disentangling Cellular Heterogeneity
Jul 02, 2023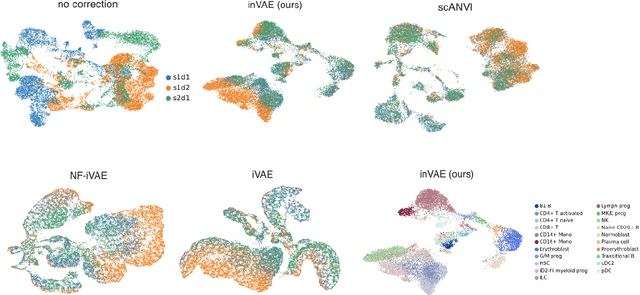
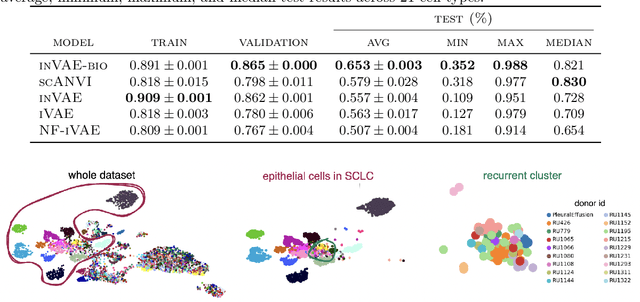
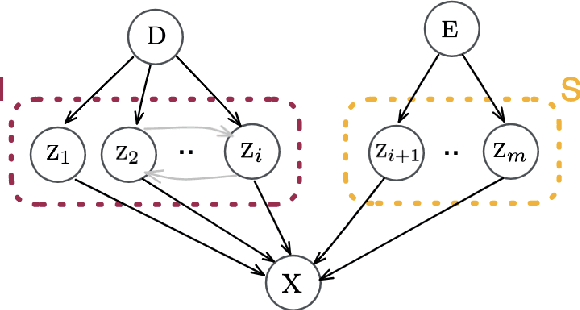
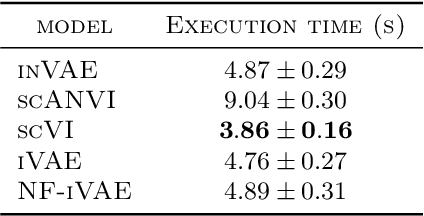
This paper presents a novel approach that leverages domain variability to learn representations that are conditionally invariant to unwanted variability or distractors. Our approach identifies both spurious and invariant latent features necessary for achieving accurate reconstruction by placing distinct conditional priors on latent features. The invariant signals are disentangled from noise by enforcing independence which facilitates the construction of an interpretable model with a causal semantic. By exploiting the interplay between data domains and labels, our method simultaneously identifies invariant features and builds invariant predictors. We apply our method to grand biological challenges, such as data integration in single-cell genomics with the aim of capturing biological variations across datasets with many samples, obtained from different conditions or multiple laboratories. Our approach allows for the incorporation of specific biological mechanisms, including gene programs, disease states, or treatment conditions into the data integration process, bridging the gap between the theoretical assumptions and real biological applications. Specifically, the proposed approach helps to disentangle biological signals from data biases that are unrelated to the target task or the causal explanation of interest. Through extensive benchmarking using large-scale human hematopoiesis and human lung cancer data, we validate the superiority of our approach over existing methods and demonstrate that it can empower deeper insights into cellular heterogeneity and the identification of disease cell states.
Training Transitive and Commutative Multimodal Transformers with LoReTTa
May 23, 2023Collecting a multimodal dataset with two paired modalities A and B or B and C is difficult in practice. Obtaining a dataset with three aligned modalities A, B, and C is even more challenging. For example, some public medical datasets have only genetic sequences and microscopic images for one patient, and only genetic sequences and radiological images for another - but no dataset includes both microscopic and radiological images for the same patient. This makes it difficult to integrate and combine all modalities into a large pre-trained neural network. We introduce LoReTTa (Linking mOdalities with a tRansitive and commutativE pre-Training sTrAtegy) to address this understudied problem. Our self-supervised framework combines causal masked modeling with the rules of commutativity and transitivity to transition within and between different modalities. Thus, it can model the relation A -> C with A -> B -> C. Given a dataset containing only the disjoint combinations (A, B) and (B, C), we show that a transformer pre-trained with LoReTTa can handle any modality combination at inference time, including the never-seen pair (A, C) and the triplet (A, B, C). We evaluate our approach on a multimodal dataset derived from MNIST containing speech, vision, and language, as well as a real-world medical dataset containing mRNA, miRNA, and RPPA samples from TCGA. Compared to traditional pre-training methods, we observe up to a 100-point reduction in perplexity for autoregressive generation tasks and up to a 15% improvement in classification accuracy for previously unseen modality pairs during the pre-training phase.