 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA scalable gene network model of regulatory dynamics in single cells
Mar 25, 2025Single-cell data provide high-dimensional measurements of the transcriptional states of cells, but extracting insights into the regulatory functions of genes, particularly identifying transcriptional mechanisms affected by biological perturbations, remains a challenge. Many perturbations induce compensatory cellular responses, making it difficult to distinguish direct from indirect effects on gene regulation. Modeling how gene regulatory functions shape the temporal dynamics of these responses is key to improving our understanding of biological perturbations. Dynamical models based on differential equations offer a principled way to capture transcriptional dynamics, but their application to single-cell data has been hindered by computational constraints, stochasticity, sparsity, and noise. Existing methods either rely on low-dimensional representations or make strong simplifying assumptions, limiting their ability to model transcriptional dynamics at scale. We introduce a Functional and Learnable model of Cell dynamicS, FLeCS, that incorporates gene network structure into coupled differential equations to model gene regulatory functions. Given (pseudo)time-series single-cell data, FLeCS accurately infers cell dynamics at scale, provides improved functional insights into transcriptional mechanisms perturbed by gene knockouts, both in myeloid differentiation and K562 Perturb-seq experiments, and simulates single-cell trajectories of A549 cells following small-molecule perturbations.
Causal machine learning for single-cell genomics
Oct 23, 2023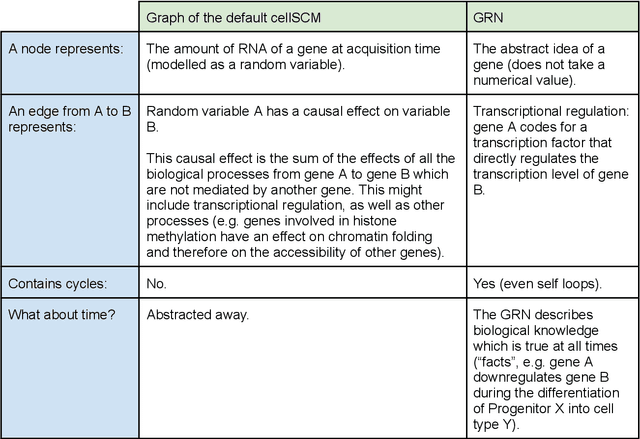
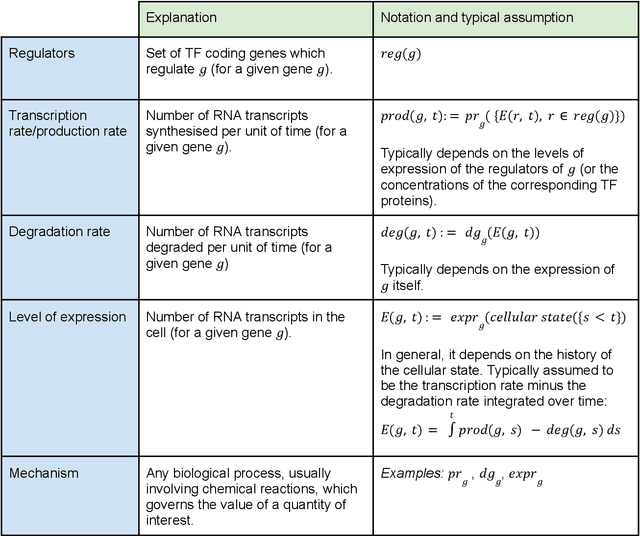
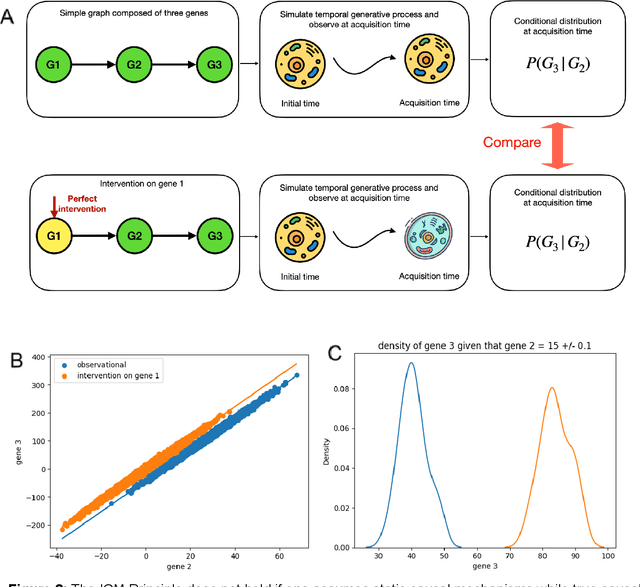

Advances in single-cell omics allow for unprecedented insights into the transcription profiles of individual cells. When combined with large-scale perturbation screens, through which specific biological mechanisms can be targeted, these technologies allow for measuring the effect of targeted perturbations on the whole transcriptome. These advances provide an opportunity to better understand the causative role of genes in complex biological processes such as gene regulation, disease progression or cellular development. However, the high-dimensional nature of the data, coupled with the intricate complexity of biological systems renders this task nontrivial. Within the machine learning community, there has been a recent increase of interest in causality, with a focus on adapting established causal techniques and algorithms to handle high-dimensional data. In this perspective, we delineate the application of these methodologies within the realm of single-cell genomics and their challenges. We first present the model that underlies most of current causal approaches to single-cell biology and discuss and challenge the assumptions it entails from the biological point of view. We then identify open problems in the application of causal approaches to single-cell data: generalising to unseen environments, learning interpretable models, and learning causal models of dynamics. For each problem, we discuss how various research directions - including the development of computational approaches and the adaptation of experimental protocols - may offer ways forward, or on the contrary pose some difficulties. With the advent of single cell atlases and increasing perturbation data, we expect causal models to become a crucial tool for informed experimental design.