 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA scalable gene network model of regulatory dynamics in single cells
Mar 25, 2025Single-cell data provide high-dimensional measurements of the transcriptional states of cells, but extracting insights into the regulatory functions of genes, particularly identifying transcriptional mechanisms affected by biological perturbations, remains a challenge. Many perturbations induce compensatory cellular responses, making it difficult to distinguish direct from indirect effects on gene regulation. Modeling how gene regulatory functions shape the temporal dynamics of these responses is key to improving our understanding of biological perturbations. Dynamical models based on differential equations offer a principled way to capture transcriptional dynamics, but their application to single-cell data has been hindered by computational constraints, stochasticity, sparsity, and noise. Existing methods either rely on low-dimensional representations or make strong simplifying assumptions, limiting their ability to model transcriptional dynamics at scale. We introduce a Functional and Learnable model of Cell dynamicS, FLeCS, that incorporates gene network structure into coupled differential equations to model gene regulatory functions. Given (pseudo)time-series single-cell data, FLeCS accurately infers cell dynamics at scale, provides improved functional insights into transcriptional mechanisms perturbed by gene knockouts, both in myeloid differentiation and K562 Perturb-seq experiments, and simulates single-cell trajectories of A549 cells following small-molecule perturbations.
Causal machine learning for single-cell genomics
Oct 23, 2023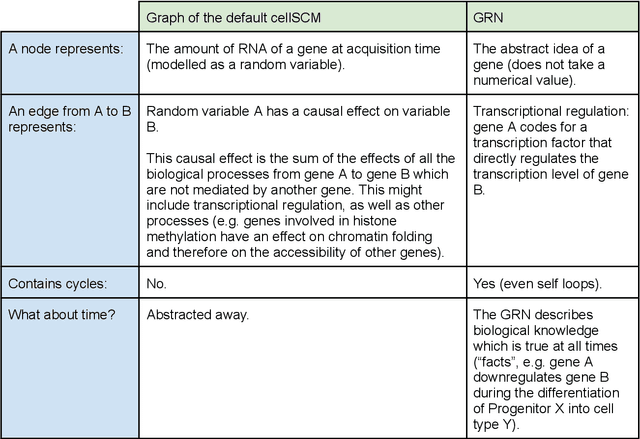
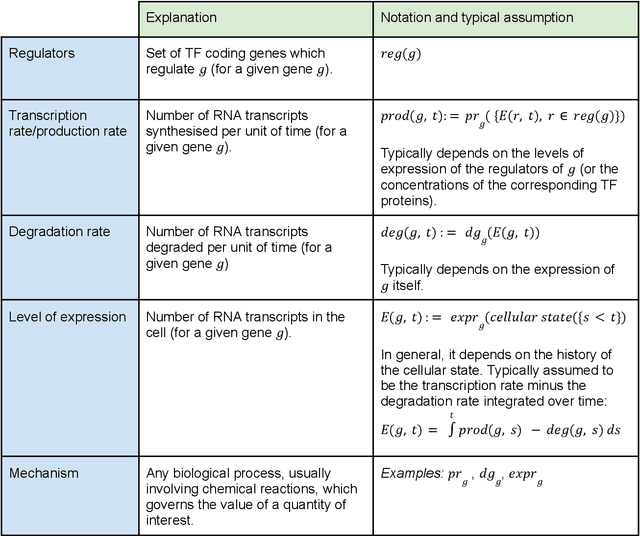
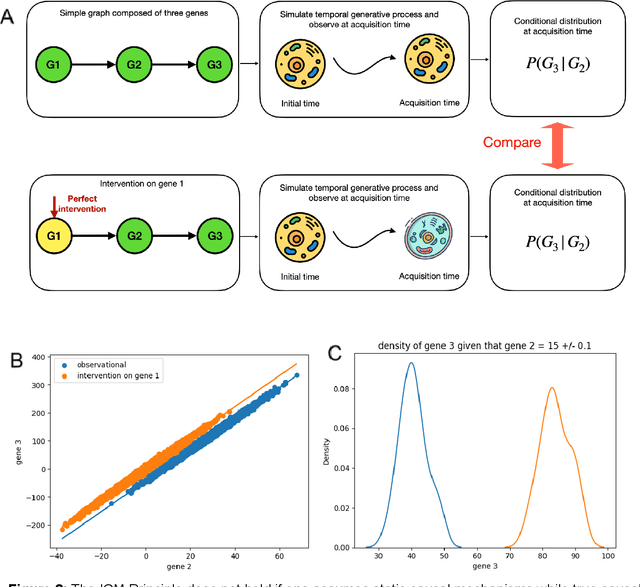

Advances in single-cell omics allow for unprecedented insights into the transcription profiles of individual cells. When combined with large-scale perturbation screens, through which specific biological mechanisms can be targeted, these technologies allow for measuring the effect of targeted perturbations on the whole transcriptome. These advances provide an opportunity to better understand the causative role of genes in complex biological processes such as gene regulation, disease progression or cellular development. However, the high-dimensional nature of the data, coupled with the intricate complexity of biological systems renders this task nontrivial. Within the machine learning community, there has been a recent increase of interest in causality, with a focus on adapting established causal techniques and algorithms to handle high-dimensional data. In this perspective, we delineate the application of these methodologies within the realm of single-cell genomics and their challenges. We first present the model that underlies most of current causal approaches to single-cell biology and discuss and challenge the assumptions it entails from the biological point of view. We then identify open problems in the application of causal approaches to single-cell data: generalising to unseen environments, learning interpretable models, and learning causal models of dynamics. For each problem, we discuss how various research directions - including the development of computational approaches and the adaptation of experimental protocols - may offer ways forward, or on the contrary pose some difficulties. With the advent of single cell atlases and increasing perturbation data, we expect causal models to become a crucial tool for informed experimental design.
RECOVER: sequential model optimization platform for combination drug repurposing identifies novel synergistic compounds in vitro
Feb 07, 2022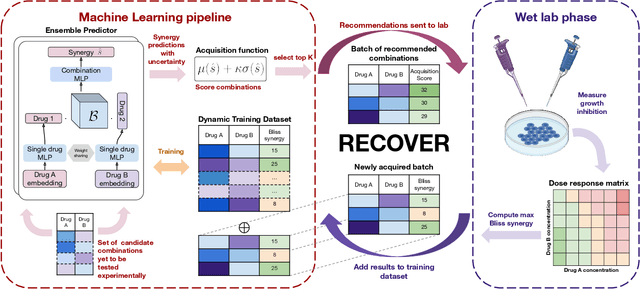

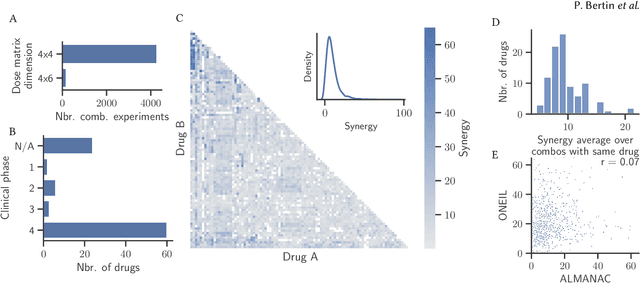
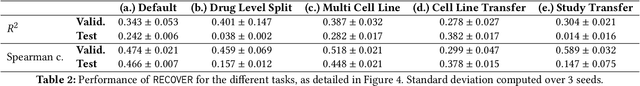
Selecting optimal drug repurposing combinations for further preclinical development is a challenging technical feat. Due to the toxicity of many therapeutic agents (e.g., chemotherapy), practitioners have favoured selection of synergistic compounds whereby lower doses can be used whilst maintaining high efficacy. For a fixed small molecule library, an exhaustive combinatorial chemical screen becomes infeasible to perform for academic and industry laboratories alike. Deep learning models have achieved state-of-the-art results in silico for the prediction of synergy scores. However, databases of drug combinations are highly biased towards synergistic agents and these results do not necessarily generalise out of distribution. We employ a sequential model optimization search applied to a deep learning model to quickly discover highly synergistic drug combinations active against a cancer cell line, while requiring substantially less screening than an exhaustive evaluation. Through iteratively adapting the model to newly acquired data, after only 3 rounds of ML-guided experimentation (including a calibration round), we find that the set of combinations queried by our model is enriched for highly synergistic combinations. Remarkably, we rediscovered a synergistic drug combination that was later confirmed to be under study within clinical trials.
TorchXRayVision: A library of chest X-ray datasets and models
Oct 31, 2021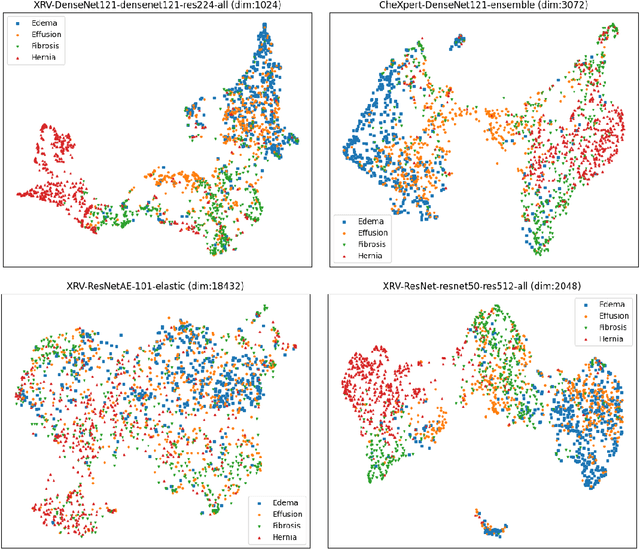
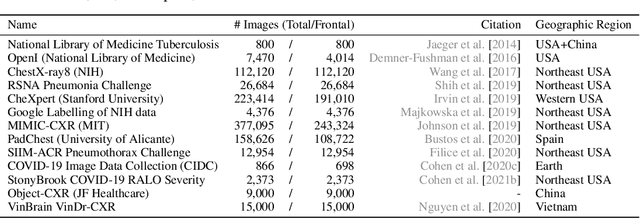
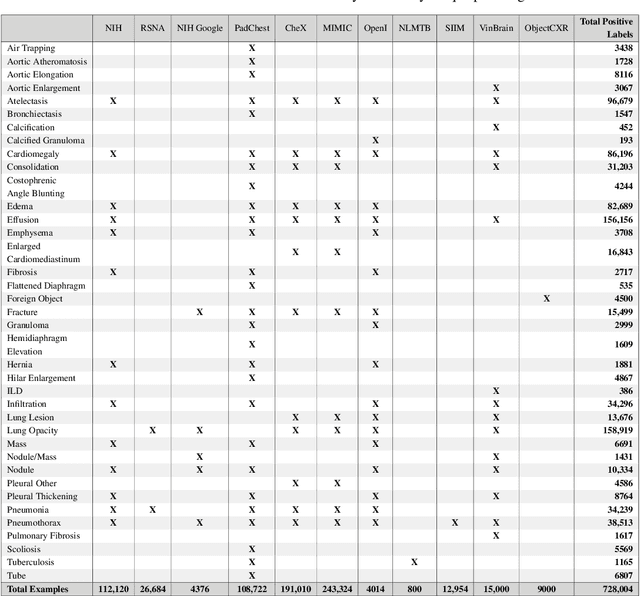
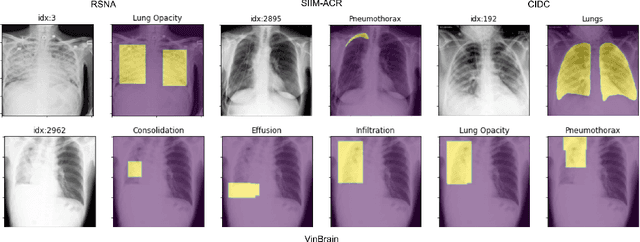
TorchXRayVision is an open source software library for working with chest X-ray datasets and deep learning models. It provides a common interface and common pre-processing chain for a wide set of publicly available chest X-ray datasets. In addition, a number of classification and representation learning models with different architectures, trained on different data combinations, are available through the library to serve as baselines or feature extractors.
DEUP: Direct Epistemic Uncertainty Prediction
Feb 16, 2021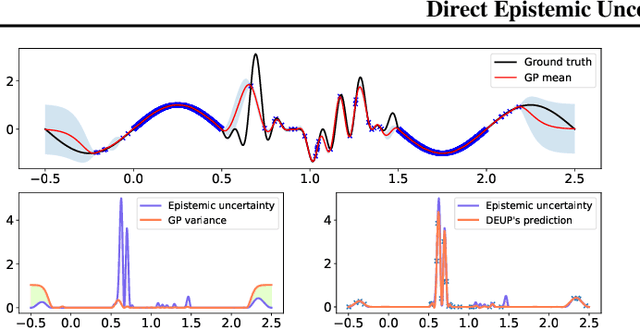
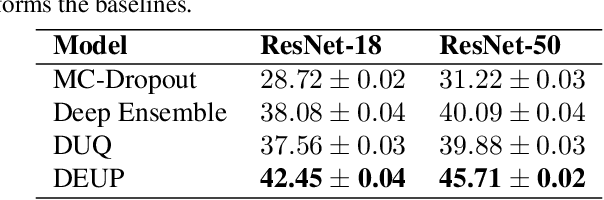
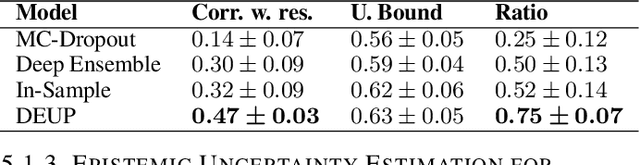
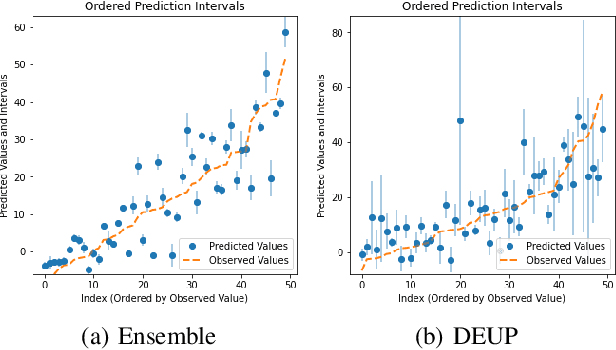
Epistemic uncertainty is the part of out-of-sample prediction error due to the lack of knowledge of the learner. Whereas previous work was focusing on model variance, we propose a principled approach for directly estimating epistemic uncertainty by learning to predict generalization error and subtracting an estimate of aleatoric uncertainty, i.e., intrinsic unpredictability. This estimator of epistemic uncertainty includes the effect of model bias and can be applied in non-stationary learning environments arising in active learning or reinforcement learning. In addition to demonstrating these properties of Direct Epistemic Uncertainty Prediction (DEUP), we illustrate its advantage against existing methods for uncertainty estimation on downstream tasks including sequential model optimization and reinforcement learning. We also evaluate the quality of uncertainty estimates from DEUP for probabilistic classification of images and for estimating uncertainty about synergistic drug combinations.
Is graph-based feature selection of genes better than random?
Nov 19, 2019
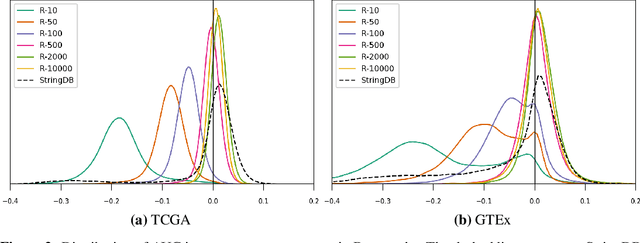
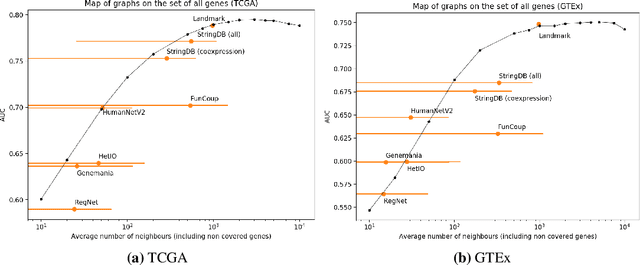
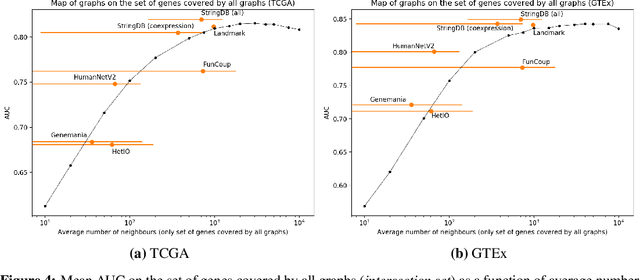
Gene interaction graphs aim to capture various relationships between genes and represent decades of biology research. When trying to make predictions from genomic data, those graphs could be used to overcome the curse of dimensionality by making machine learning models sparser and more consistent with biological common knowledge. In this work, we focus on assessing whether those graphs capture dependencies seen in gene expression data better than random. We formulate a condition that graphs should satisfy to provide a good prior knowledge and propose to test it using a `Single Gene Inference' (SGI) task. We compare random graphs with seven major gene interaction graphs published by different research groups, aiming to measure the true benefit of using biologically relevant graphs in this context. Our analysis finds that dependencies can be captured almost as well at random which suggests that, in terms of gene expression levels, the relevant information about the state of the cell is spread across many genes.
Analysis of Gene Interaction Graphs for Biasing Machine Learning Models
May 06, 2019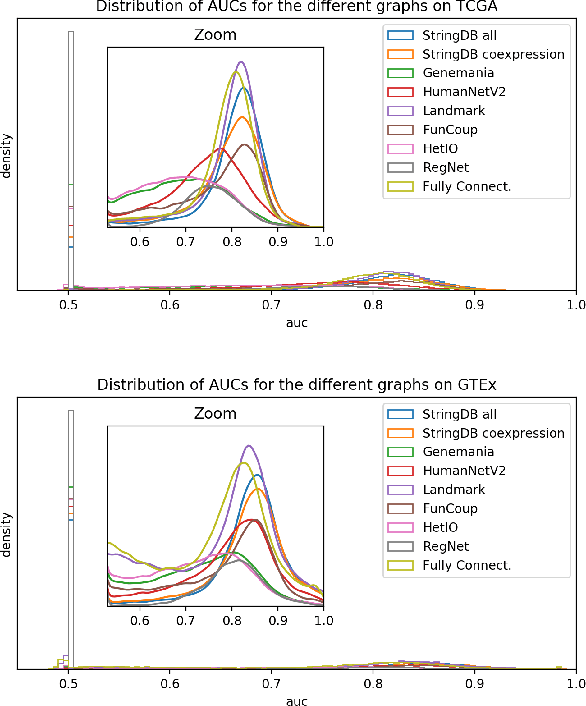
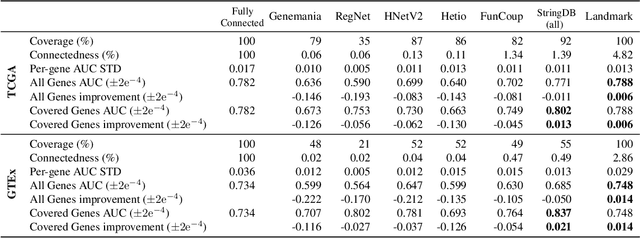
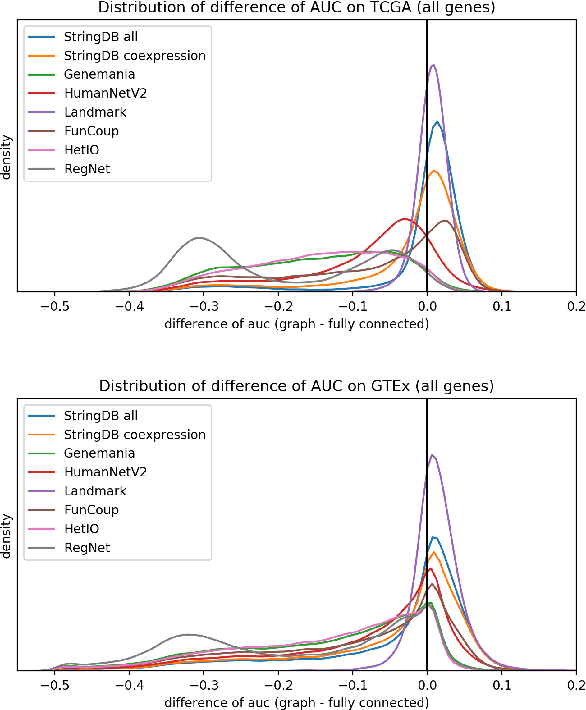
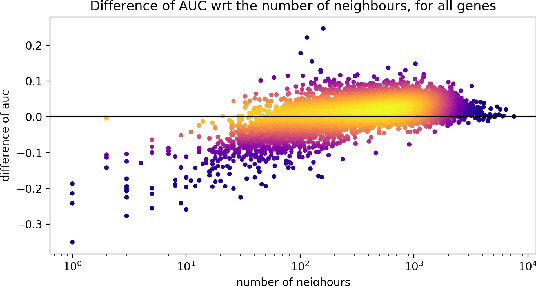
Gene interaction graphs aim to capture various relationships between genes and can be used to create more biologically-intuitive models for machine learning. There are many such graphs available which can differ in the number of genes and edges covered. In this work, we attempt to evaluate the biases provided by those graphs through utilizing them for 'Single Gene Inference' (SGI) which serves as, what we believe is, a proxy for more relevant prediction tasks. The SGI task assesses how well a gene's neighbors in a particular graph can 'explain' the gene itself in comparison to the baseline of using all the genes in the dataset. We evaluate seven major gene interaction graphs created by different research groups on two distinct datasets, TCGA and GTEx. We find that some graphs perform on par with the unbiased baseline for most genes with a significantly smaller feature set.
Chester: A Web Delivered Locally Computed Chest X-Ray Disease Prediction System
Jan 31, 2019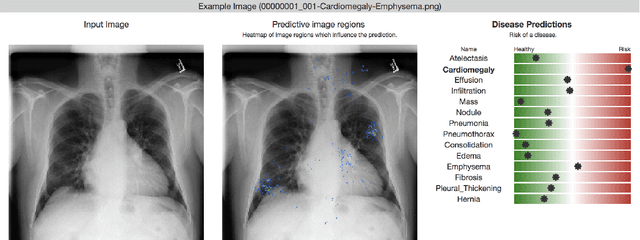
Deep learning has shown promise to augment radiologists and improve the standard of care globally. Two main issues that complicate deploying these systems are patient privacy and scaling to the global population. To deploy a system at scale with minimal computational cost while preserving privacy we present a web delivered (but locally run) system for diagnosing chest X-Rays. Code is delivered via a URL to a web browser (including cell phones) but the patient data remains on the users machine and all processing occurs locally. The system is designed to be used as a reference where a user can process an image to confirm or aid in their diagnosis. The system contains three main components: out-of-distribution detection, disease prediction, and prediction explanation. The system open source and freely available here: https://mlmed.org/tools/xray/