 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTorchXRayVision: A library of chest X-ray datasets and models
Oct 31, 2021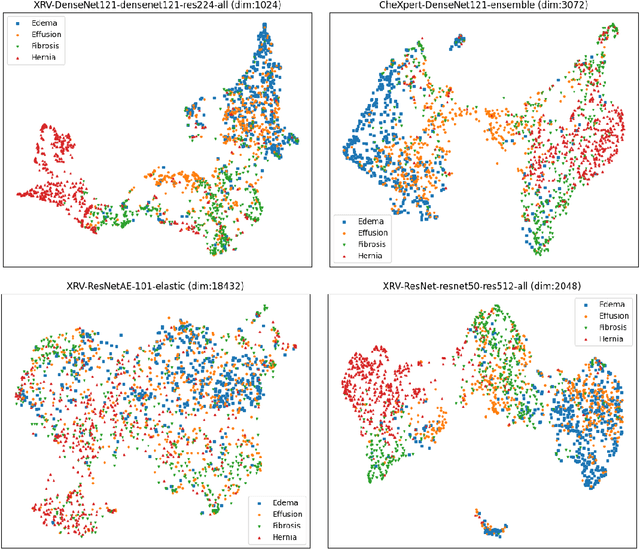
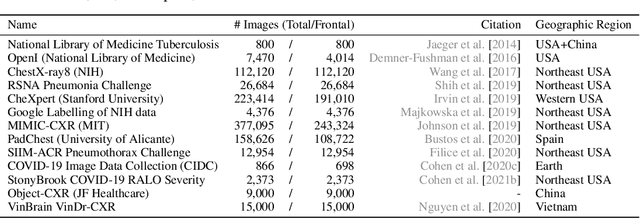
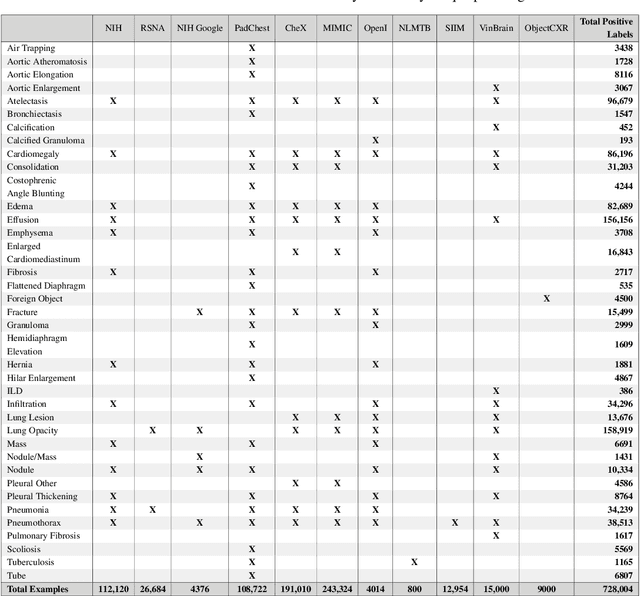
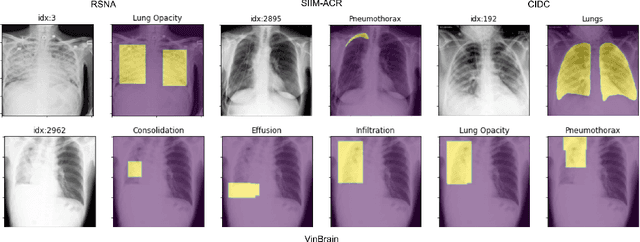
TorchXRayVision is an open source software library for working with chest X-ray datasets and deep learning models. It provides a common interface and common pre-processing chain for a wide set of publicly available chest X-ray datasets. In addition, a number of classification and representation learning models with different architectures, trained on different data combinations, are available through the library to serve as baselines or feature extractors.
Quantifying the Value of Lateral Views in Deep Learning for Chest X-rays
Feb 07, 2020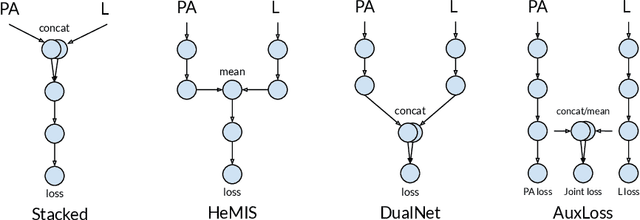
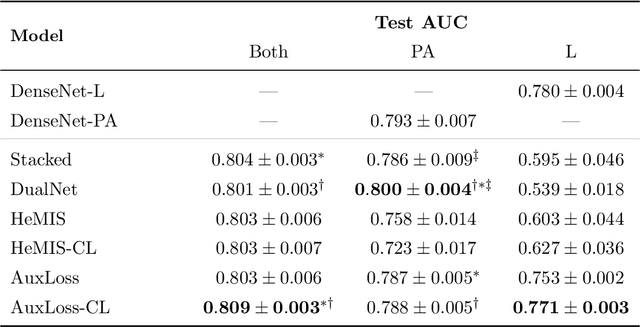
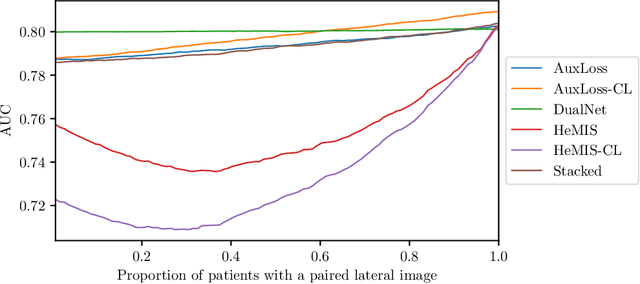
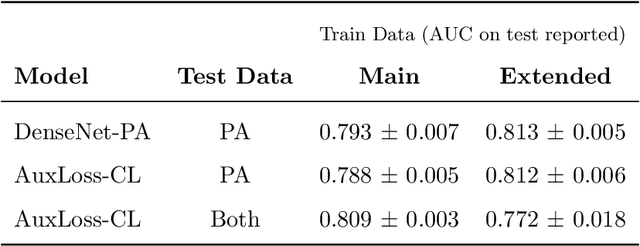
Most deep learning models in chest X-ray prediction utilize the posteroanterior (PA) view due to the lack of other views available. PadChest is a large-scale chest X-ray dataset that has almost 200 labels and multiple views available. In this work, we use PadChest to explore multiple approaches to merging the PA and lateral views for predicting the radiological labels associated with the X-ray image. We find that different methods of merging the model utilize the lateral view differently. We also find that including the lateral view increases performance for 32 labels in the dataset, while being neutral for the others. The increase in overall performance is comparable to the one obtained by using only the PA view with twice the amount of patients in the training set.
On the limits of cross-domain generalization in automated X-ray prediction
Feb 06, 2020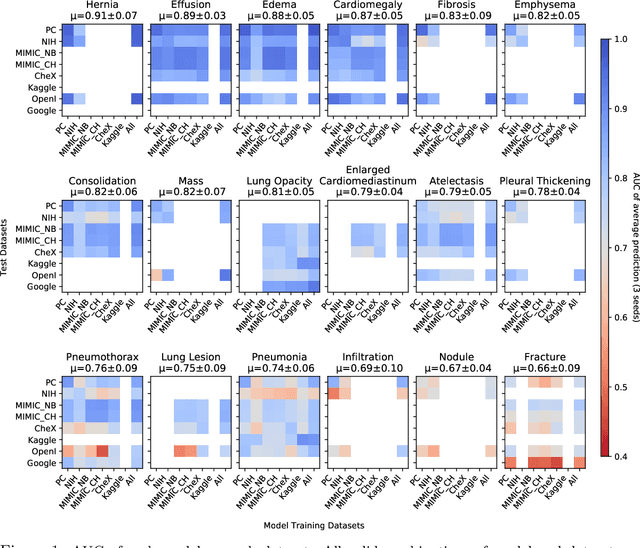
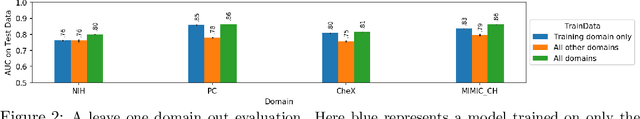
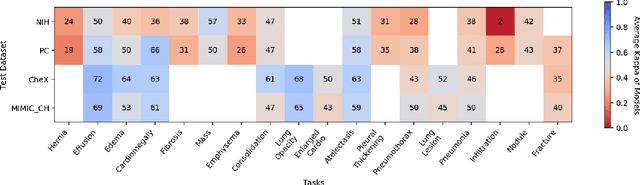
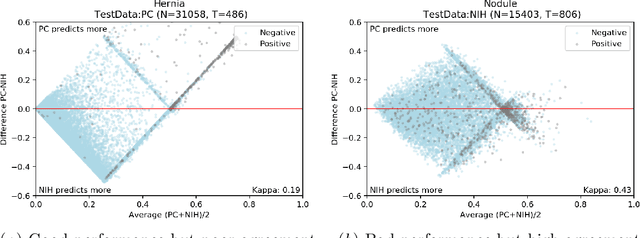
This large scale study focuses on quantifying what X-rays diagnostic prediction tasks generalize well across multiple different datasets. We present evidence that the issue of generalization is not due to a shift in the images but instead a shift in the labels. We study the cross-domain performance, agreement between models, and model representations. We find interesting discrepancies between performance and agreement where models which both achieve good performance disagree in their predictions as well as models which agree yet achieve poor performance. We also test for concept similarity by regularizing a network to group tasks across multiple datasets together and observe variation across the tasks.
Is graph-based feature selection of genes better than random?
Nov 19, 2019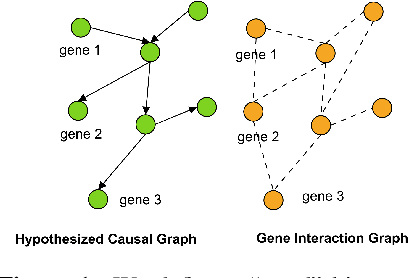
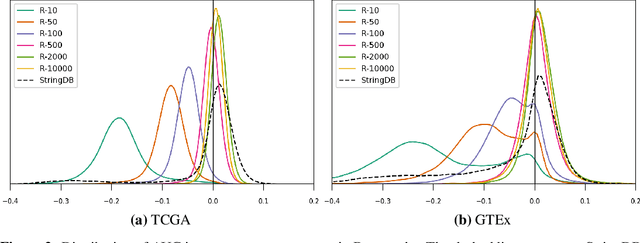
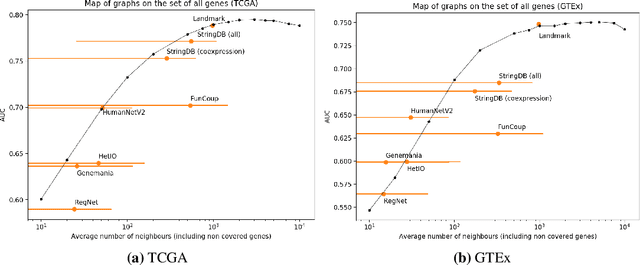
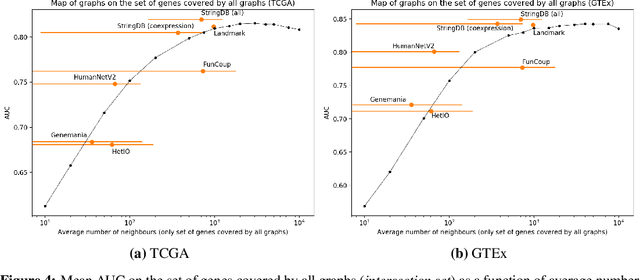
Gene interaction graphs aim to capture various relationships between genes and represent decades of biology research. When trying to make predictions from genomic data, those graphs could be used to overcome the curse of dimensionality by making machine learning models sparser and more consistent with biological common knowledge. In this work, we focus on assessing whether those graphs capture dependencies seen in gene expression data better than random. We formulate a condition that graphs should satisfy to provide a good prior knowledge and propose to test it using a `Single Gene Inference' (SGI) task. We compare random graphs with seven major gene interaction graphs published by different research groups, aiming to measure the true benefit of using biologically relevant graphs in this context. Our analysis finds that dependencies can be captured almost as well at random which suggests that, in terms of gene expression levels, the relevant information about the state of the cell is spread across many genes.
Towards unstructured mortality prediction with free-text clinical notes
Nov 19, 2019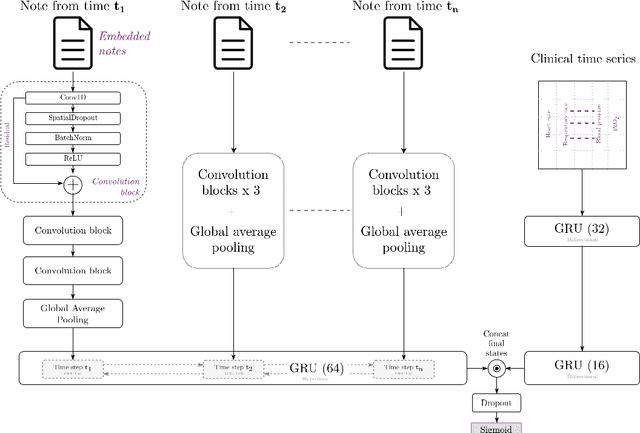
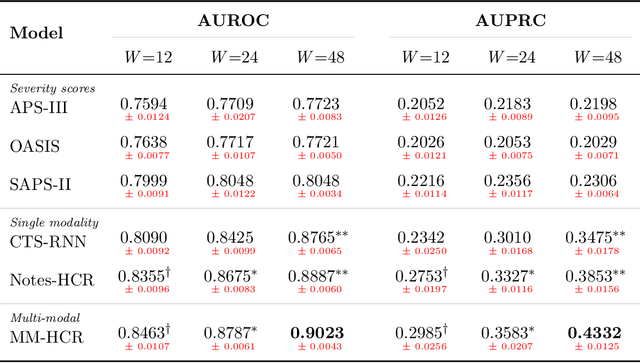
Healthcare data continues to flourish yet a relatively small portion, mostly structured, is being utilized effectively for predicting clinical outcomes. The rich subjective information available in unstructured clinical notes can possibly facilitate higher discrimination but tends to be under-utilized in mortality prediction. This work attempts to assess the gain in performance when multiple notes that have been minimally preprocessed are used as an input for prediction. A hierarchical architecture consisting of both convolutional and recurrent layers is used to concurrently model the different notes compiled in an individual hospital stay. This approach is evaluated on predicting in-hospital mortality on the MIMIC-III dataset. On comparison to approaches utilizing structured data, it achieved higher metrics despite requiring less cleaning and preprocessing. This demonstrates the potential of unstructured data in enhancing mortality prediction and signifies the need to incorporate more raw unstructured data into current clinical prediction methods.
Analysis of Gene Interaction Graphs for Biasing Machine Learning Models
May 06, 2019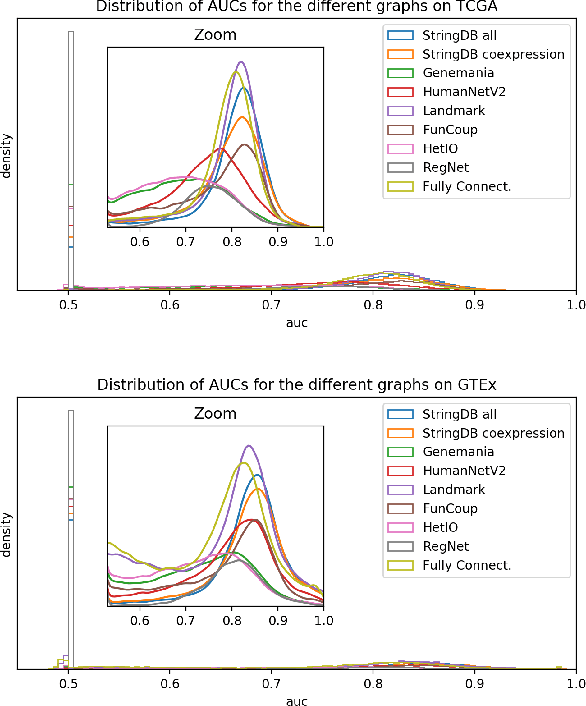
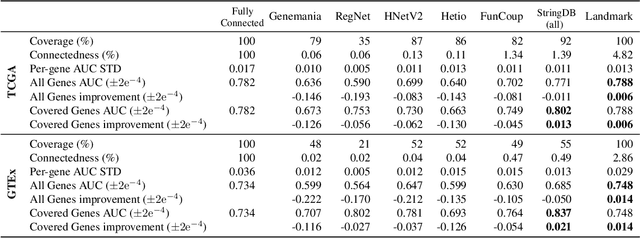
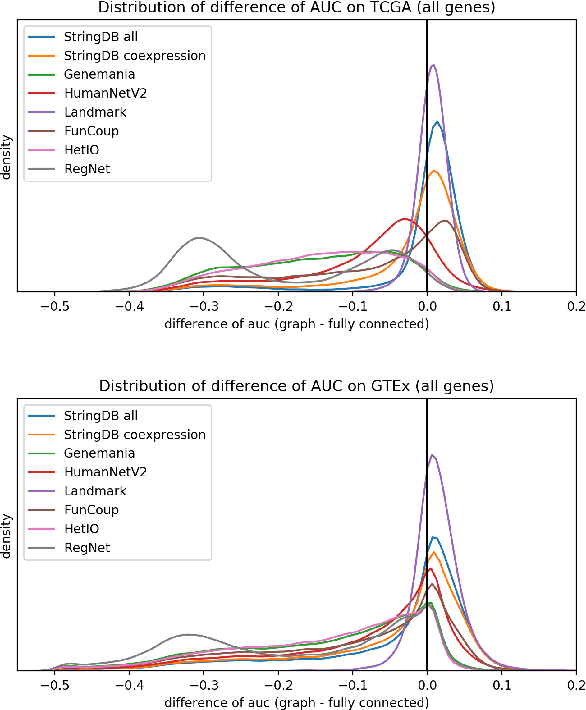
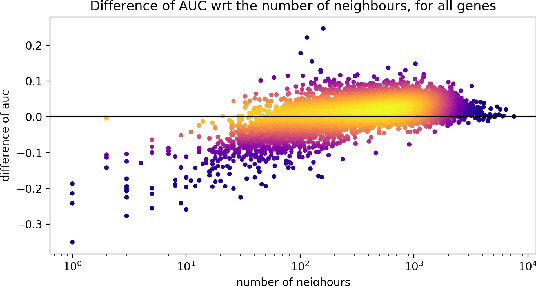
Gene interaction graphs aim to capture various relationships between genes and can be used to create more biologically-intuitive models for machine learning. There are many such graphs available which can differ in the number of genes and edges covered. In this work, we attempt to evaluate the biases provided by those graphs through utilizing them for 'Single Gene Inference' (SGI) which serves as, what we believe is, a proxy for more relevant prediction tasks. The SGI task assesses how well a gene's neighbors in a particular graph can 'explain' the gene itself in comparison to the baseline of using all the genes in the dataset. We evaluate seven major gene interaction graphs created by different research groups on two distinct datasets, TCGA and GTEx. We find that some graphs perform on par with the unbiased baseline for most genes with a significantly smaller feature set.
Do Lateral Views Help Automated Chest X-ray Predictions?
Apr 17, 2019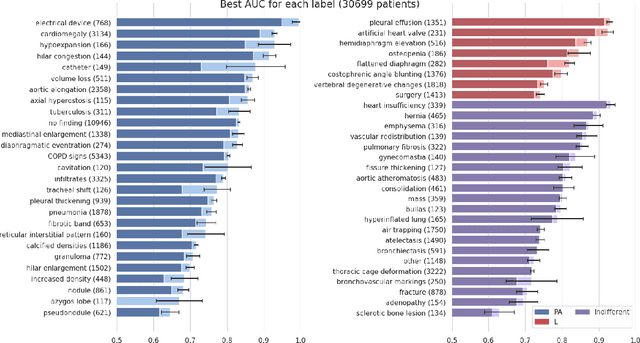
Most convolutional neural networks in chest radiology use only the frontal posteroanterior (PA) view to make a prediction. However the lateral view is known to help the diagnosis of certain diseases and conditions. The recently released PadChest dataset contains paired PA and lateral views, allowing us to study for which diseases and conditions the performance of a neural network improves when provided a lateral x-ray view as opposed to a frontal posteroanterior (PA) view. Using a simple DenseNet model, we find that using the lateral view increases the AUC of 8 of the 56 labels in our data and achieves the same performance as the PA view for 21 of the labels. We find that using the PA and lateral views jointly doesn't trivially lead to an increase in performance but suggest further investigation.