 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeIs graph-based feature selection of genes better than random?
Nov 19, 2019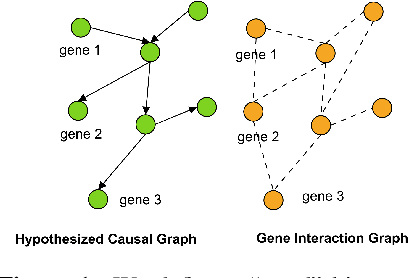
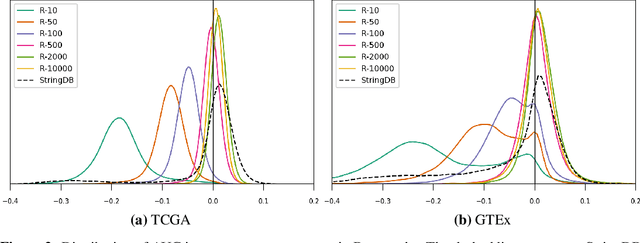
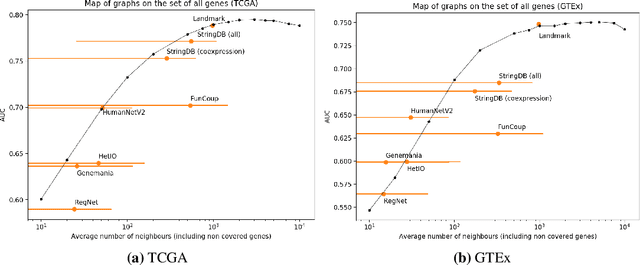
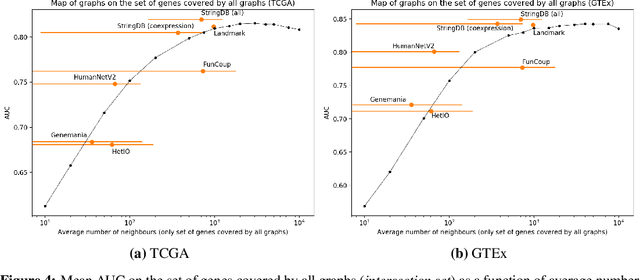
Gene interaction graphs aim to capture various relationships between genes and represent decades of biology research. When trying to make predictions from genomic data, those graphs could be used to overcome the curse of dimensionality by making machine learning models sparser and more consistent with biological common knowledge. In this work, we focus on assessing whether those graphs capture dependencies seen in gene expression data better than random. We formulate a condition that graphs should satisfy to provide a good prior knowledge and propose to test it using a `Single Gene Inference' (SGI) task. We compare random graphs with seven major gene interaction graphs published by different research groups, aiming to measure the true benefit of using biologically relevant graphs in this context. Our analysis finds that dependencies can be captured almost as well at random which suggests that, in terms of gene expression levels, the relevant information about the state of the cell is spread across many genes.
The TCGA Meta-Dataset Clinical Benchmark
Oct 18, 2019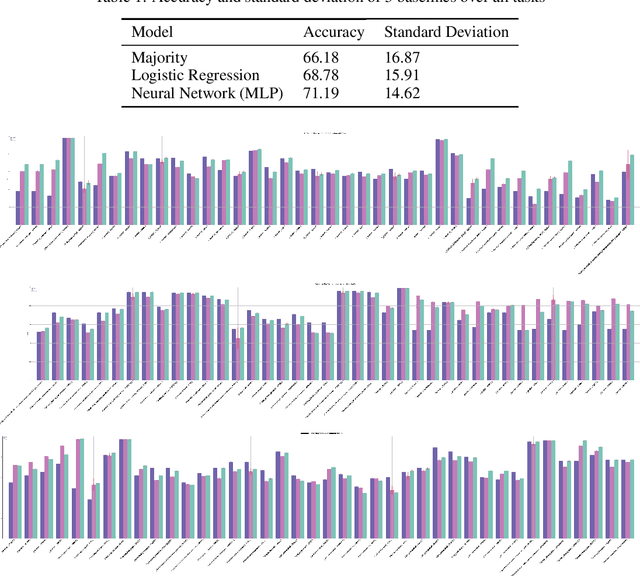
Machine learning is bringing a paradigm shift to healthcare by changing the process of disease diagnosis and prognosis in clinics and hospitals. This development equips doctors and medical staff with tools to evaluate their hypotheses and hence make more precise decisions. Although most current research in the literature seeks to develop techniques and methods for predicting one particular clinical outcome, this approach is far from the reality of clinical decision making in which you have to consider several factors simultaneously. In addition, it is difficult to follow the recent progress concretely as there is a lack of consistency in benchmark datasets and task definitions in the field of Genomics. To address the aforementioned issues, we provide a clinical Meta-Dataset derived from the publicly available data hub called The Cancer Genome Atlas Program (TCGA) that contains 174 tasks. We believe those tasks could be good proxy tasks to develop methods which can work on a few samples of gene expression data. Also, learning to predict multiple clinical variables using gene-expression data is an important task due to the variety of phenotypes in clinical problems and lack of samples for some of the rare variables. The defined tasks cover a wide range of clinical problems including predicting tumor tissue site, white cell count, histological type, family history of cancer, gender, and many others which we explain later in the paper. Each task represents an independent dataset. We use regression and neural network baselines for all the tasks using only 150 samples and compare their performance.
Analysis of Gene Interaction Graphs for Biasing Machine Learning Models
May 06, 2019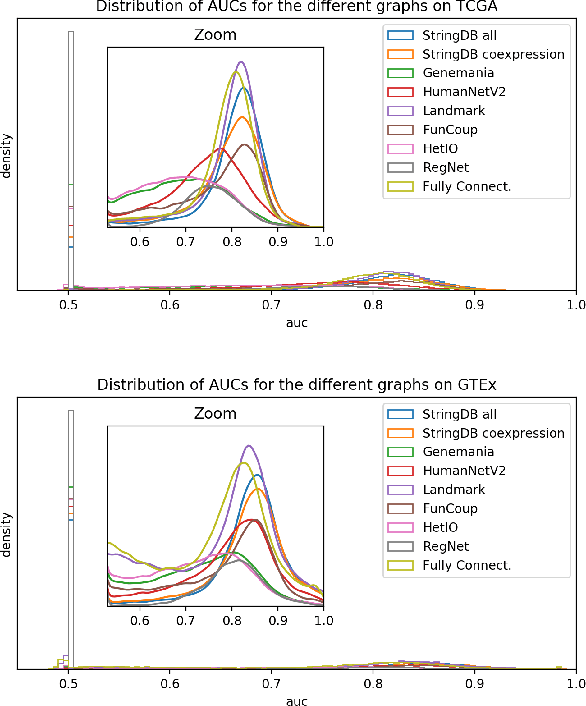
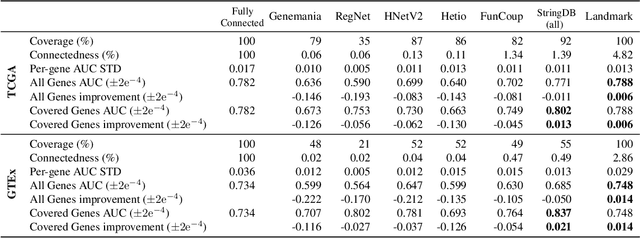
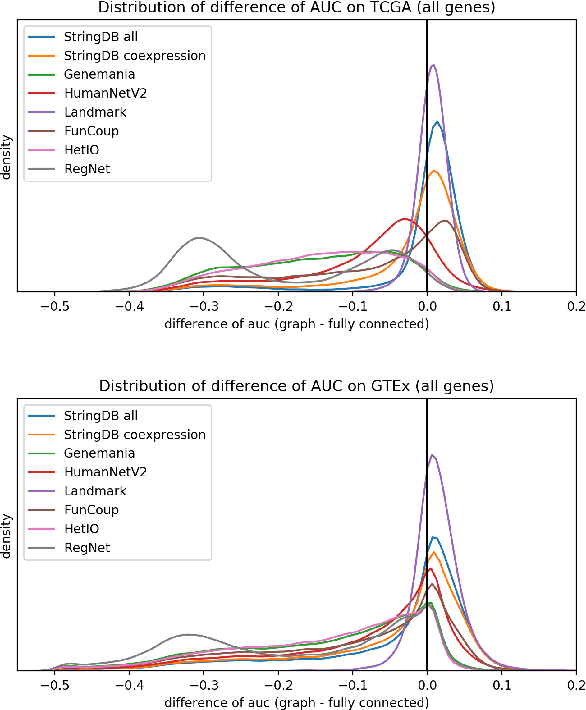
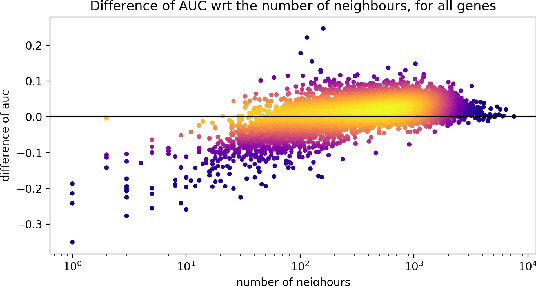
Gene interaction graphs aim to capture various relationships between genes and can be used to create more biologically-intuitive models for machine learning. There are many such graphs available which can differ in the number of genes and edges covered. In this work, we attempt to evaluate the biases provided by those graphs through utilizing them for 'Single Gene Inference' (SGI) which serves as, what we believe is, a proxy for more relevant prediction tasks. The SGI task assesses how well a gene's neighbors in a particular graph can 'explain' the gene itself in comparison to the baseline of using all the genes in the dataset. We evaluate seven major gene interaction graphs created by different research groups on two distinct datasets, TCGA and GTEx. We find that some graphs perform on par with the unbiased baseline for most genes with a significantly smaller feature set.