 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeThe TCGA Meta-Dataset Clinical Benchmark
Oct 18, 2019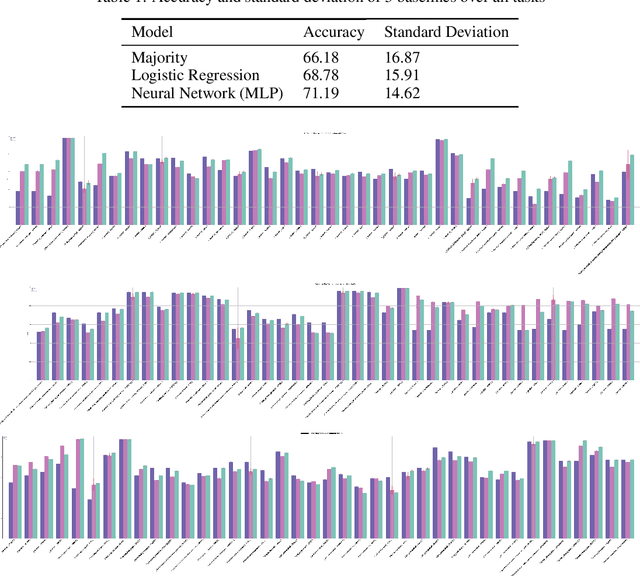
Machine learning is bringing a paradigm shift to healthcare by changing the process of disease diagnosis and prognosis in clinics and hospitals. This development equips doctors and medical staff with tools to evaluate their hypotheses and hence make more precise decisions. Although most current research in the literature seeks to develop techniques and methods for predicting one particular clinical outcome, this approach is far from the reality of clinical decision making in which you have to consider several factors simultaneously. In addition, it is difficult to follow the recent progress concretely as there is a lack of consistency in benchmark datasets and task definitions in the field of Genomics. To address the aforementioned issues, we provide a clinical Meta-Dataset derived from the publicly available data hub called The Cancer Genome Atlas Program (TCGA) that contains 174 tasks. We believe those tasks could be good proxy tasks to develop methods which can work on a few samples of gene expression data. Also, learning to predict multiple clinical variables using gene-expression data is an important task due to the variety of phenotypes in clinical problems and lack of samples for some of the rare variables. The defined tasks cover a wide range of clinical problems including predicting tumor tissue site, white cell count, histological type, family history of cancer, gender, and many others which we explain later in the paper. Each task represents an independent dataset. We use regression and neural network baselines for all the tasks using only 150 samples and compare their performance.
Towards the Latent Transcriptome
Oct 08, 2018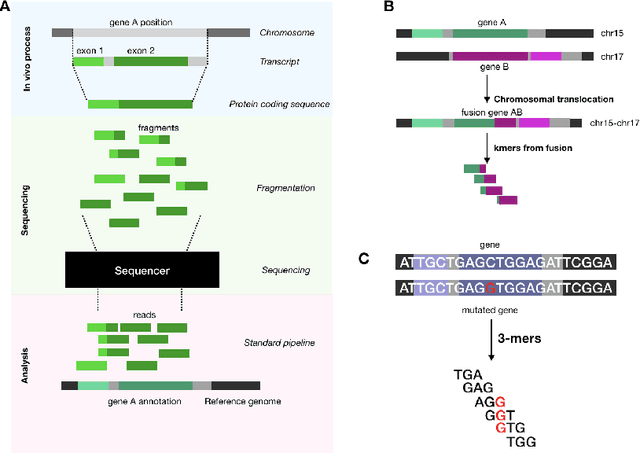

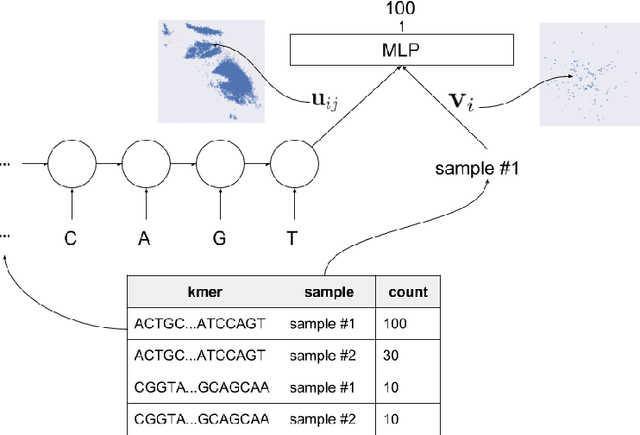
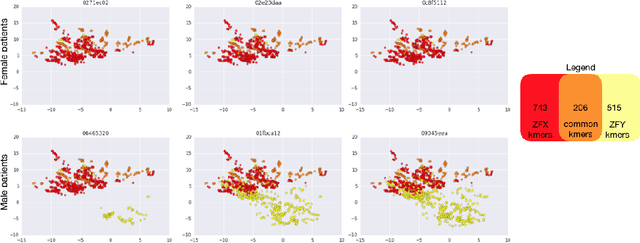
In this work we propose a method to compute continuous embeddings for kmers from raw RNA-seq data, in a reference-free fashion. We report that our model captures information of both DNA sequence similarity as well as DNA sequence abundance in the embedding latent space. We confirm the quality of these vectors by comparing them to known gene sub-structures and report that the latent space recovers exon information from raw RNA-Seq data from acute myeloid leukemia patients. Furthermore we show that this latent space allows the detection of genomic abnormalities such as translocations as well as patient-specific mutations, making this representation space both useful for visualization as well as analysis.