 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA multimodal and temporal foundation model for virtual patient representations at healthcare system scale
Apr 21, 2026Modern medicine generates vast multimodal data across siloed systems, yet no existing model integrates the full breadth and temporal depth of the clinical record into a unified patient representation. We introduce Apollo, a multimodal temporal foundation model trained and evaluated on over three decades of longitudinal hospital records from a major US hospital system, composed of 25 billion records from 7.2 million patients, representing 28 distinct medical modalities and 12 major medical specialties. Apollo learns a unified representation space integrating over 100 thousand unique medical events in our clinical vocabulary as well as images and clinical text. This "atlas of medical concepts" forms a computational substrate for modeling entire patient care journeys comprised of sequences of structured and unstructured events, which are compressed by Apollo into virtual patient representations. To assess the potential of these whole-patient representations, we created 322 prognosis and retrieval tasks from a held-out test set of 1.4 million patients. We demonstrate the generalized clinical forecasting potential of Apollo embeddings, including predicting new disease onset risk up to five years in advance (95 tasks), disease progression (78 tasks), treatment response (59 tasks), risk of treatment-related adverse events (17 tasks), and hospital operations endpoints (12 tasks). Using feature attribution techniques, we show that model predictions align with clinically-interpretable multimodal biomarkers. We evaluate semantic similarity search on 61 retrieval tasks, and moreover demonstrate the potential of Apollo as a multimodal medical search engine using text and image queries. Together, these modeling capabilities establish the foundation for computable medicine, where the full context of patient care becomes accessible to computational reasoning.
Towards Spatial Transcriptomics-driven Pathology Foundation Models
Feb 15, 2026Spatial transcriptomics (ST) provides spatially resolved measurements of gene expression, enabling characterization of the molecular landscape of human tissue beyond histological assessment as well as localized readouts that can be aligned with morphology. Concurrently, the success of multimodal foundation models that integrate vision with complementary modalities suggests that morphomolecular coupling between local expression and morphology can be systematically used to improve histological representations themselves. We introduce Spatial Expression-Aligned Learning (SEAL), a vision-omics self-supervised learning framework that infuses localized molecular information into pathology vision encoders. Rather than training new encoders from scratch, SEAL is designed as a parameter-efficient vision-omics finetuning method that can be flexibly applied to widely used pathology foundation models. We instantiate SEAL by training on over 700,000 paired gene expression spot-tissue region examples spanning tumor and normal samples from 14 organs. Tested across 38 slide-level and 15 patch-level downstream tasks, SEAL provides a drop-in replacement for pathology foundation models that consistently improves performance over widely used vision-only and ST prediction baselines on slide-level molecular status, pathway activity, and treatment response prediction, as well as patch-level gene expression prediction tasks. Additionally, SEAL encoders exhibit robust domain generalization on out-of-distribution evaluations and enable new cross-modal capabilities such as gene-to-image retrieval. Our work proposes a general framework for ST-guided finetuning of pathology foundation models, showing that augmenting existing models with localized molecular supervision is an effective and practical step for improving visual representations and expanding their cross-modal utility.
Molecular-driven Foundation Model for Oncologic Pathology
Jan 28, 2025Foundation models are reshaping computational pathology by enabling transfer learning, where models pre-trained on vast datasets can be adapted for downstream diagnostic, prognostic, and therapeutic response tasks. Despite these advances, foundation models are still limited in their ability to encode the entire gigapixel whole-slide images without additional training and often lack complementary multimodal data. Here, we introduce Threads, a slide-level foundation model capable of generating universal representations of whole-slide images of any size. Threads was pre-trained using a multimodal learning approach on a diverse cohort of 47,171 hematoxylin and eosin (H&E)-stained tissue sections, paired with corresponding genomic and transcriptomic profiles - the largest such paired dataset to be used for foundation model development to date. This unique training paradigm enables Threads to capture the tissue's underlying molecular composition, yielding powerful representations applicable to a wide array of downstream tasks. In extensive benchmarking across 54 oncology tasks, including clinical subtyping, grading, mutation prediction, immunohistochemistry status determination, treatment response prediction, and survival prediction, Threads outperformed all baselines while demonstrating remarkable generalizability and label efficiency. It is particularly well suited for predicting rare events, further emphasizing its clinical utility. We intend to make the model publicly available for the broader community.
Multimodal Whole Slide Foundation Model for Pathology
Nov 29, 2024



The field of computational pathology has been transformed with recent advances in foundation models that encode histopathology region-of-interests (ROIs) into versatile and transferable feature representations via self-supervised learning (SSL). However, translating these advancements to address complex clinical challenges at the patient and slide level remains constrained by limited clinical data in disease-specific cohorts, especially for rare clinical conditions. We propose TITAN, a multimodal whole slide foundation model pretrained using 335,645 WSIs via visual self-supervised learning and vision-language alignment with corresponding pathology reports and 423,122 synthetic captions generated from a multimodal generative AI copilot for pathology. Without any finetuning or requiring clinical labels, TITAN can extract general-purpose slide representations and generate pathology reports that generalize to resource-limited clinical scenarios such as rare disease retrieval and cancer prognosis. We evaluate TITAN on diverse clinical tasks and find that TITAN outperforms both ROI and slide foundation models across machine learning settings such as linear probing, few-shot and zero-shot classification, rare cancer retrieval and cross-modal retrieval, and pathology report generation.
HEST-1k: A Dataset for Spatial Transcriptomics and Histology Image Analysis
Jun 23, 2024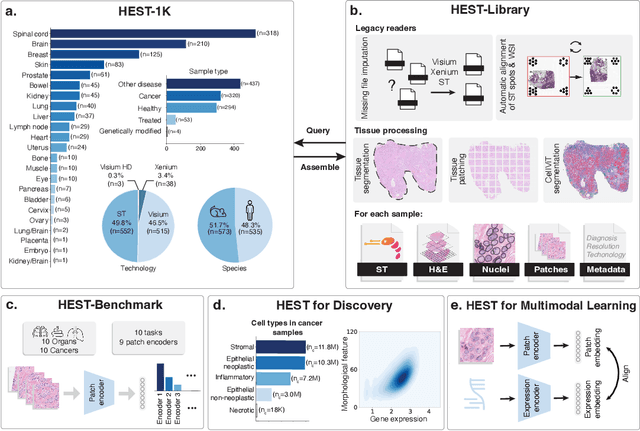
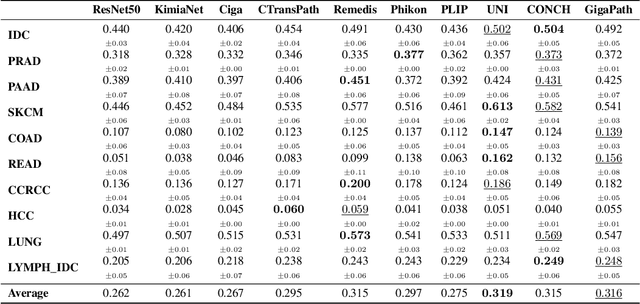
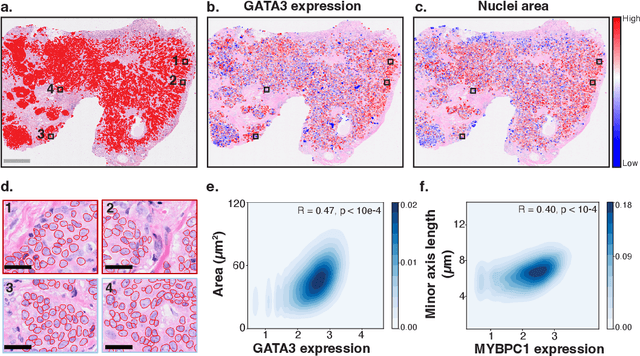
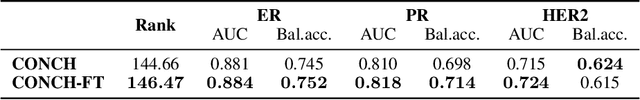
Spatial transcriptomics (ST) enables interrogating the molecular composition of tissue with ever-increasing resolution, depth, and sensitivity. However, costs, rapidly evolving technology, and lack of standards have constrained computational methods in ST to narrow tasks and small cohorts. In addition, the underlying tissue morphology as reflected by H&E-stained whole slide images (WSIs) encodes rich information often overlooked in ST studies. Here, we introduce HEST-1k, a collection of 1,108 spatial transcriptomic profiles, each linked to a WSI and metadata. HEST-1k was assembled using HEST-Library from 131 public and internal cohorts encompassing 25 organs, two species (Homo Sapiens and Mus Musculus), and 320 cancer samples from 25 cancer types. HEST-1k processing enabled the identification of 1.5 million expression--morphology pairs and 60 million nuclei. HEST-1k is tested on three use cases: (1) benchmarking foundation models for histopathology (HEST-Benchmark), (2) biomarker identification, and (3) multimodal representation learning. HEST-1k, HEST-Library, and HEST-Benchmark can be freely accessed via https://github.com/mahmoodlab/hest.
DinoBloom: A Foundation Model for Generalizable Cell Embeddings in Hematology
Apr 07, 2024In hematology, computational models offer significant potential to improve diagnostic accuracy, streamline workflows, and reduce the tedious work of analyzing single cells in peripheral blood or bone marrow smears. However, clinical adoption of computational models has been hampered by the lack of generalization due to large batch effects, small dataset sizes, and poor performance in transfer learning from natural images. To address these challenges, we introduce DinoBloom, the first foundation model for single cell images in hematology, utilizing a tailored DINOv2 pipeline. Our model is built upon an extensive collection of 13 diverse, publicly available datasets of peripheral blood and bone marrow smears, the most substantial open-source cohort in hematology so far, comprising over 380,000 white blood cell images. To assess its generalization capability, we evaluate it on an external dataset with a challenging domain shift. We show that our model outperforms existing medical and non-medical vision models in (i) linear probing and k-nearest neighbor evaluations for cell-type classification on blood and bone marrow smears and (ii) weakly supervised multiple instance learning for acute myeloid leukemia subtyping by a large margin. A family of four DinoBloom models (small, base, large, and giant) can be adapted for a wide range of downstream applications, be a strong baseline for classification problems, and facilitate the assessment of batch effects in new datasets. All models are available at github.com/marrlab/DinoBloom.
B-Cos Aligned Transformers Learn Human-Interpretable Features
Jan 18, 2024Vision Transformers (ViTs) and Swin Transformers (Swin) are currently state-of-the-art in computational pathology. However, domain experts are still reluctant to use these models due to their lack of interpretability. This is not surprising, as critical decisions need to be transparent and understandable. The most common approach to understanding transformers is to visualize their attention. However, attention maps of ViTs are often fragmented, leading to unsatisfactory explanations. Here, we introduce a novel architecture called the B-cos Vision Transformer (BvT) that is designed to be more interpretable. It replaces all linear transformations with the B-cos transform to promote weight-input alignment. In a blinded study, medical experts clearly ranked BvTs above ViTs, suggesting that our network is better at capturing biomedically relevant structures. This is also true for the B-cos Swin Transformer (Bwin). Compared to the Swin Transformer, it even improves the F1-score by up to 4.7% on two public datasets.
Low-resource finetuning of foundation models beats state-of-the-art in histopathology
Jan 09, 2024To handle the large scale of whole slide images in computational pathology, most approaches first tessellate the images into smaller patches, extract features from these patches, and finally aggregate the feature vectors with weakly-supervised learning. The performance of this workflow strongly depends on the quality of the extracted features. Recently, foundation models in computer vision showed that leveraging huge amounts of data through supervised or self-supervised learning improves feature quality and generalizability for a variety of tasks. In this study, we benchmark the most popular vision foundation models as feature extractors for histopathology data. We evaluate the models in two settings: slide-level classification and patch-level classification. We show that foundation models are a strong baseline. Our experiments demonstrate that by finetuning a foundation model on a single GPU for only two hours or three days depending on the dataset, we can match or outperform state-of-the-art feature extractors for computational pathology. These findings imply that even with little resources one can finetune a feature extractor tailored towards a specific downstream task and dataset. This is a considerable shift from the current state, where only few institutions with large amounts of resources and datasets are able to train a feature extractor. We publish all code used for training and evaluation as well as the finetuned models.
From Whole-slide Image to Biomarker Prediction: A Protocol for End-to-End Deep Learning in Computational Pathology
Dec 18, 2023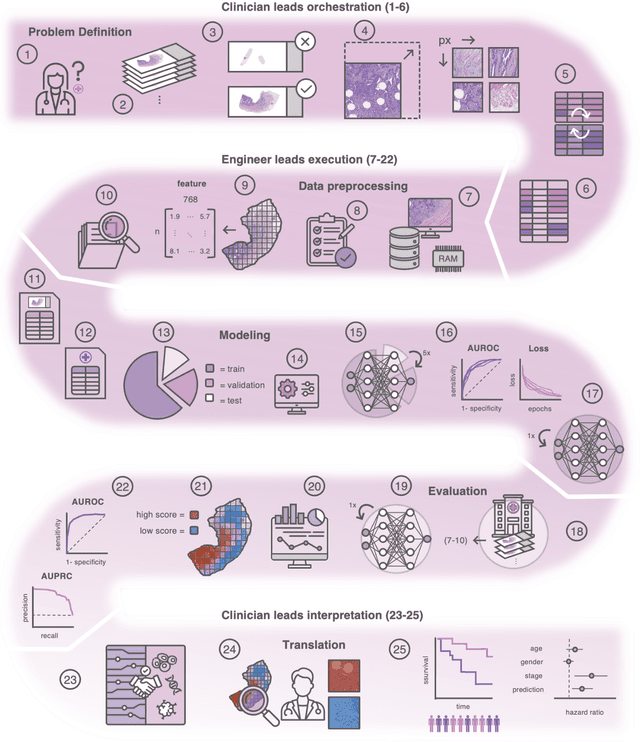
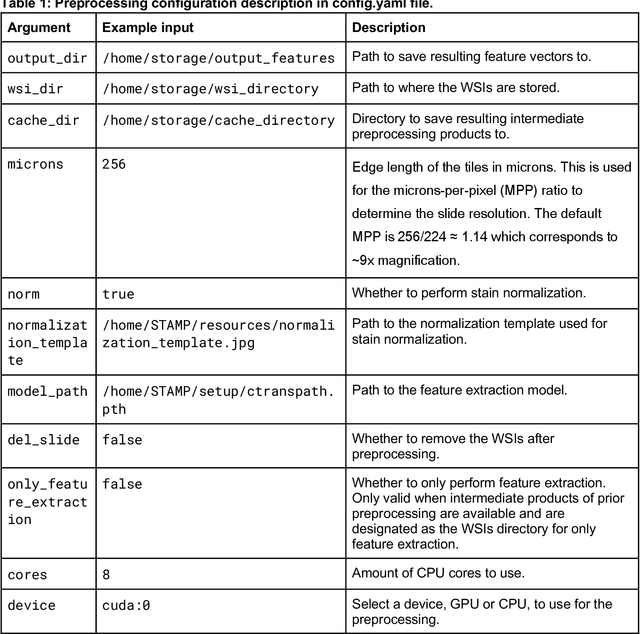
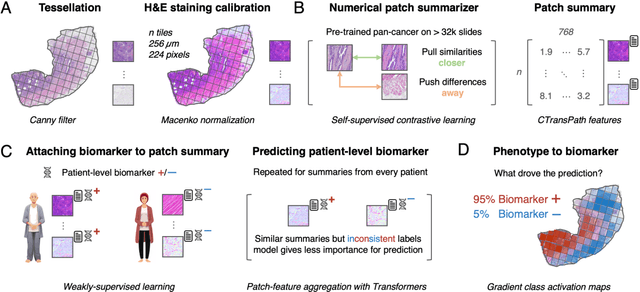

Hematoxylin- and eosin (H&E) stained whole-slide images (WSIs) are the foundation of diagnosis of cancer. In recent years, development of deep learning-based methods in computational pathology enabled the prediction of biomarkers directly from WSIs. However, accurately linking tissue phenotype to biomarkers at scale remains a crucial challenge for democratizing complex biomarkers in precision oncology. This protocol describes a practical workflow for solid tumor associative modeling in pathology (STAMP), enabling prediction of biomarkers directly from WSIs using deep learning. The STAMP workflow is biomarker agnostic and allows for genetic- and clinicopathologic tabular data to be included as an additional input, together with histopathology images. The protocol consists of five main stages which have been successfully applied to various research problems: formal problem definition, data preprocessing, modeling, evaluation and clinical translation. The STAMP workflow differentiates itself through its focus on serving as a collaborative framework that can be used by clinicians and engineers alike for setting up research projects in the field of computational pathology. As an example task, we applied STAMP to the prediction of microsatellite instability (MSI) status in colorectal cancer, showing accurate performance for the identification of MSI-high tumors. Moreover, we provide an open-source codebase which has been deployed at several hospitals across the globe to set up computational pathology workflows. The STAMP workflow requires one workday of hands-on computational execution and basic command line knowledge.
Fully transformer-based biomarker prediction from colorectal cancer histology: a large-scale multicentric study
Jan 23, 2023



Background: Deep learning (DL) can extract predictive and prognostic biomarkers from routine pathology slides in colorectal cancer. For example, a DL test for the diagnosis of microsatellite instability (MSI) in CRC has been approved in 2022. Current approaches rely on convolutional neural networks (CNNs). Transformer networks are outperforming CNNs and are replacing them in many applications, but have not been used for biomarker prediction in cancer at a large scale. In addition, most DL approaches have been trained on small patient cohorts, which limits their clinical utility. Methods: In this study, we developed a new fully transformer-based pipeline for end-to-end biomarker prediction from pathology slides. We combine a pre-trained transformer encoder and a transformer network for patch aggregation, capable of yielding single and multi-target prediction at patient level. We train our pipeline on over 9,000 patients from 10 colorectal cancer cohorts. Results: A fully transformer-based approach massively improves the performance, generalizability, data efficiency, and interpretability as compared with current state-of-the-art algorithms. After training on a large multicenter cohort, we achieve a sensitivity of 0.97 with a negative predictive value of 0.99 for MSI prediction on surgical resection specimens. We demonstrate for the first time that resection specimen-only training reaches clinical-grade performance on endoscopic biopsy tissue, solving a long-standing diagnostic problem. Interpretation: A fully transformer-based end-to-end pipeline trained on thousands of pathology slides yields clinical-grade performance for biomarker prediction on surgical resections and biopsies. Our new methods are freely available under an open source license.