 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeUncertainty-guided Generation of Dark-field Radiographs
Jan 22, 2026X-ray dark-field radiography provides complementary diagnostic information to conventional attenuation imaging by visualizing microstructural tissue changes through small-angle scattering. However, the limited availability of such data poses challenges for developing robust deep learning models. In this work, we present the first framework for generating dark-field images directly from standard attenuation chest X-rays using an Uncertainty-Guided Progressive Generative Adversarial Network. The model incorporates both aleatoric and epistemic uncertainty to improve interpretability and reliability. Experiments demonstrate high structural fidelity of the generated images, with consistent improvement of quantitative metrics across stages. Furthermore, out-of-distribution evaluation confirms that the proposed model generalizes well. Our results indicate that uncertainty-guided generative modeling enables realistic dark-field image synthesis and provides a reliable foundation for future clinical applications.
No Data? No Problem: Robust Vision-Tabular Learning with Missing Values
Dec 22, 2025



Large-scale medical biobanks provide imaging data complemented by extensive tabular information, such as demographics or clinical measurements. However, this abundance of tabular attributes does not reflect real-world datasets, where only a subset of attributes may be available. This discrepancy calls for methods that can leverage all the tabular data during training while remaining robust to missing values at inference. To address this challenge, we propose RoVTL (Robust Vision-Tabular Learning), a framework designed to handle any level of tabular data availability, from 0% to 100%. RoVTL comprises two key stages: contrastive pretraining, where we introduce tabular attribute missingness as data augmentation to promote robustness, and downstream task tuning using a gated cross-attention module for multimodal fusion. During fine-tuning, we employ a novel Tabular More vs. Fewer loss that ranks performance based on the amount of available tabular data. Combined with disentangled gradient learning, this enables consistent performance across all tabular data completeness scenarios. We evaluate RoVTL on cardiac MRI scans from the UK Biobank, demonstrating superior robustness to missing tabular data compared to prior methods. Furthermore, RoVTL successfully generalizes to an external cardiac MRI dataset for multimodal disease classification, and extends to the natural images domain, achieving robust performance on a car advertisements dataset. The code is available at https://github.com/marteczkah/RoVTL.
TGV: Tabular Data-Guided Learning of Visual Cardiac Representations
Mar 19, 2025Contrastive learning methods in computer vision typically rely on different views of the same image to form pairs. However, in medical imaging, we often seek to compare entire patients with different phenotypes rather than just multiple augmentations of one scan. We propose harnessing clinically relevant tabular data to identify distinct patient phenotypes and form more meaningful pairs in a contrastive learning framework. Our method uses tabular attributes to guide the training of visual representations, without requiring a joint embedding space. We demonstrate its strength using short-axis cardiac MR images and clinical attributes from the UK Biobank, where tabular data helps to more effectively distinguish between patient subgroups. Evaluation on downstream tasks, including fine-tuning and zero-shot prediction of cardiovascular artery diseases and cardiac phenotypes, shows that incorporating tabular data yields stronger visual representations than conventional methods that rely solely on image augmentations or combined image-tabular embeddings. Furthermore, we demonstrate that image encoders trained with tabular guidance are capable of embedding demographic information in their representations, allowing them to use insights from tabular data for unimodal predictions, making them well-suited to real-world medical settings where extensive clinical annotations may not be routinely available at inference time. The code will be available on GitHub.
Subspace Implicit Neural Representations for Real-Time Cardiac Cine MR Imaging
Dec 17, 2024
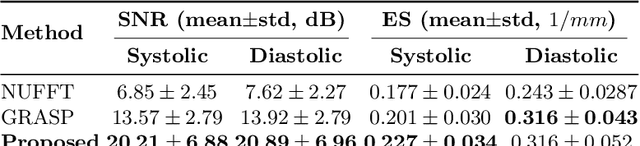
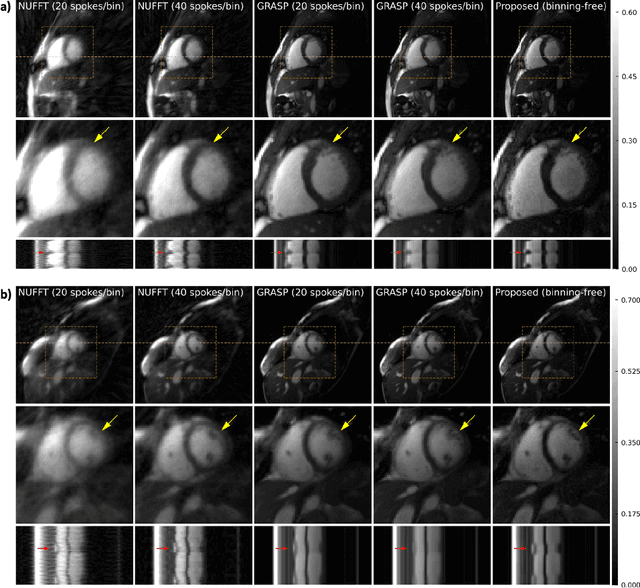
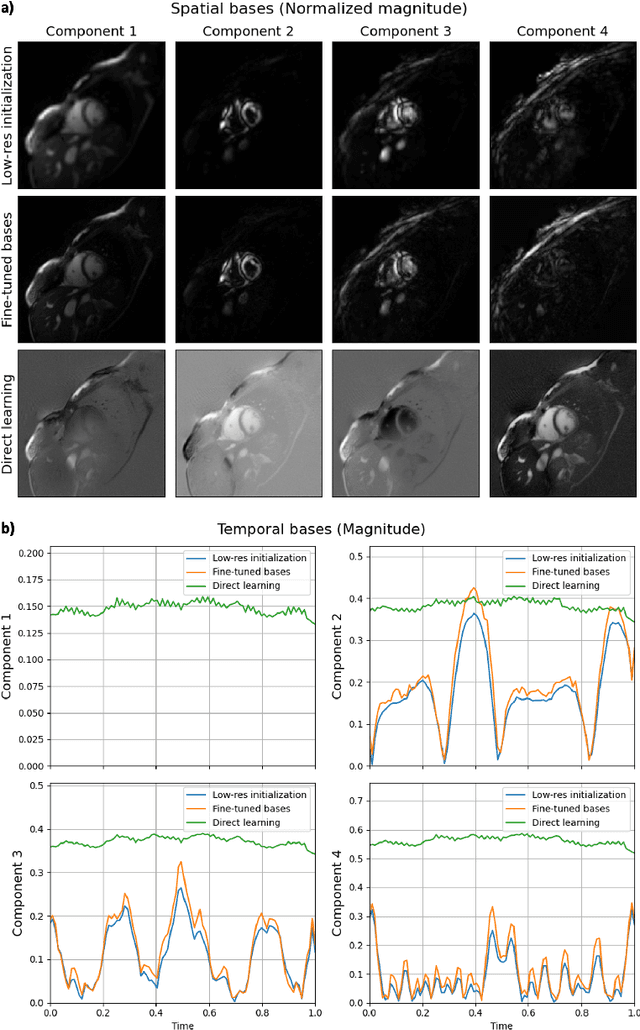
Conventional cardiac cine MRI methods rely on retrospective gating, which limits temporal resolution and the ability to capture continuous cardiac dynamics, particularly in patients with arrhythmias and beat-to-beat variations. To address these challenges, we propose a reconstruction framework based on subspace implicit neural representations for real-time cardiac cine MRI of continuously sampled radial data. This approach employs two multilayer perceptrons to learn spatial and temporal subspace bases, leveraging the low-rank properties of cardiac cine MRI. Initialized with low-resolution reconstructions, the networks are fine-tuned using spoke-specific loss functions to recover spatial details and temporal fidelity. Our method directly utilizes the continuously sampled radial k-space spokes during training, thereby eliminating the need for binning and non-uniform FFT. This approach achieves superior spatial and temporal image quality compared to conventional binned methods at the acceleration rate of 10 and 20, demonstrating potential for high-resolution imaging of dynamic cardiac events and enhancing diagnostic capability.
A Self-Supervised Image Registration Approach for Measuring Local Response Patterns in Metastatic Ovarian Cancer
Jul 24, 2024



High-grade serous ovarian carcinoma (HGSOC) is characterised by significant spatial and temporal heterogeneity, typically manifesting at an advanced metastatic stage. A major challenge in treating advanced HGSOC is effectively monitoring localised change in tumour burden across multiple sites during neoadjuvant chemotherapy (NACT) and predicting long-term pathological response and overall patient survival. In this work, we propose a self-supervised deformable image registration algorithm that utilises a general-purpose image encoder for image feature extraction to co-register contrast-enhanced computerised tomography scan images acquired before and after neoadjuvant chemotherapy. This approach addresses challenges posed by highly complex tumour deformations and longitudinal lesion matching during treatment. Localised tumour changes are calculated using the Jacobian determinant maps of the registration deformation at multiple disease sites and their macroscopic areas, including hypo-dense (i.e., cystic/necrotic), hyper-dense (i.e., calcified), and intermediate density (i.e., soft tissue) portions. A series of experiments is conducted to understand the role of a general-purpose image encoder and its application in quantifying change in tumour burden during neoadjuvant chemotherapy in HGSOC. This work is the first to demonstrate the feasibility of a self-supervised image registration approach in quantifying NACT-induced localised tumour changes across the whole disease burden of patients with complex multi-site HGSOC, which could be used as a potential marker for ovarian cancer patient's long-term pathological response and survival.
DinoBloom: A Foundation Model for Generalizable Cell Embeddings in Hematology
Apr 07, 2024In hematology, computational models offer significant potential to improve diagnostic accuracy, streamline workflows, and reduce the tedious work of analyzing single cells in peripheral blood or bone marrow smears. However, clinical adoption of computational models has been hampered by the lack of generalization due to large batch effects, small dataset sizes, and poor performance in transfer learning from natural images. To address these challenges, we introduce DinoBloom, the first foundation model for single cell images in hematology, utilizing a tailored DINOv2 pipeline. Our model is built upon an extensive collection of 13 diverse, publicly available datasets of peripheral blood and bone marrow smears, the most substantial open-source cohort in hematology so far, comprising over 380,000 white blood cell images. To assess its generalization capability, we evaluate it on an external dataset with a challenging domain shift. We show that our model outperforms existing medical and non-medical vision models in (i) linear probing and k-nearest neighbor evaluations for cell-type classification on blood and bone marrow smears and (ii) weakly supervised multiple instance learning for acute myeloid leukemia subtyping by a large margin. A family of four DinoBloom models (small, base, large, and giant) can be adapted for a wide range of downstream applications, be a strong baseline for classification problems, and facilitate the assessment of batch effects in new datasets. All models are available at github.com/marrlab/DinoBloom.
Towards Learning Contrast Kinetics with Multi-Condition Latent Diffusion Models
Mar 20, 2024



Contrast agents in dynamic contrast enhanced magnetic resonance imaging allow to localize tumors and observe their contrast kinetics, which is essential for cancer characterization and respective treatment decision-making. However, contrast agent administration is not only associated with adverse health risks, but also restricted for patients during pregnancy, and for those with kidney malfunction, or other adverse reactions. With contrast uptake as key biomarker for lesion malignancy, cancer recurrence risk, and treatment response, it becomes pivotal to reduce the dependency on intravenous contrast agent administration. To this end, we propose a multi-conditional latent diffusion model capable of acquisition time-conditioned image synthesis of DCE-MRI temporal sequences. To evaluate medical image synthesis, we additionally propose and validate the Fr\'echet radiomics distance as an image quality measure based on biomarker variability between synthetic and real imaging data. Our results demonstrate our method's ability to generate realistic multi-sequence fat-saturated breast DCE-MRI and uncover the emerging potential of deep learning based contrast kinetics simulation. We publicly share our accessible codebase at https://github.com/RichardObi/ccnet.
Sparse annotation strategies for segmentation of short axis cardiac MRI
Jul 24, 2023Short axis cardiac MRI segmentation is a well-researched topic, with excellent results achieved by state-of-the-art models in a supervised setting. However, annotating MRI volumes is time-consuming and expensive. Many different approaches (e.g. transfer learning, data augmentation, few-shot learning, etc.) have emerged in an effort to use fewer annotated data and still achieve similar performance as a fully supervised model. Nevertheless, to the best of our knowledge, none of these works focus on which slices of MRI volumes are most important to annotate for yielding the best segmentation results. In this paper, we investigate the effects of training with sparse volumes, i.e. reducing the number of cases annotated, and sparse annotations, i.e. reducing the number of slices annotated per case. We evaluate the segmentation performance using the state-of-the-art nnU-Net model on two public datasets to identify which slices are the most important to annotate. We have shown that training on a significantly reduced dataset (48 annotated volumes) can give a Dice score greater than 0.85 and results comparable to using the full dataset (160 and 240 volumes for each dataset respectively). In general, training on more slice annotations provides more valuable information compared to training on more volumes. Further, annotating slices from the middle of volumes yields the most beneficial results in terms of segmentation performance, and the apical region the worst. When evaluating the trade-off between annotating volumes against slices, annotating as many slices as possible instead of annotating more volumes is a better strategy.
Is MC Dropout Bayesian?
Oct 08, 2021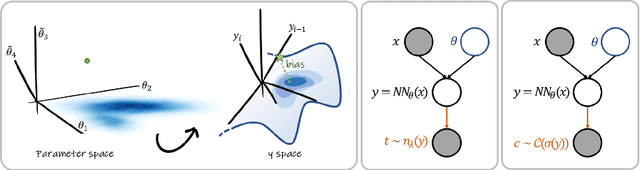
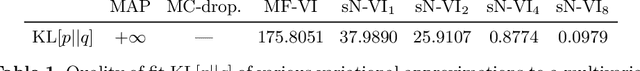
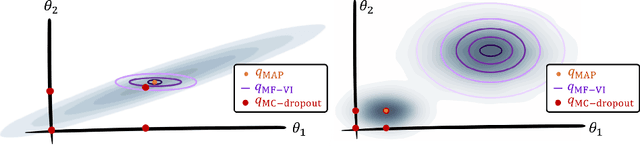

MC Dropout is a mainstream "free lunch" method in medical imaging for approximate Bayesian computations (ABC). Its appeal is to solve out-of-the-box the daunting task of ABC and uncertainty quantification in Neural Networks (NNs); to fall within the variational inference (VI) framework; and to propose a highly multimodal, faithful predictive posterior. We question the properties of MC Dropout for approximate inference, as in fact MC Dropout changes the Bayesian model; its predictive posterior assigns $0$ probability to the true model on closed-form benchmarks; the multimodality of its predictive posterior is not a property of the true predictive posterior but a design artefact. To address the need for VI on arbitrary models, we share a generic VI engine within the pytorch framework. The code includes a carefully designed implementation of structured (diagonal plus low-rank) multivariate normal variational families, and mixtures thereof. It is intended as a go-to no-free-lunch approach, addressing shortcomings of mean-field VI with an adjustable trade-off between expressivity and computational complexity.
Bayesian analysis of the prevalence bias: learning and predicting from imbalanced data
Jul 31, 2021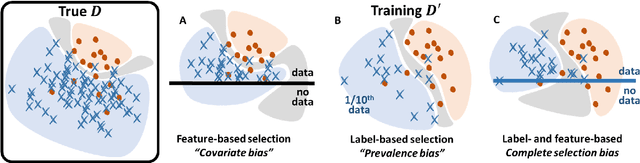
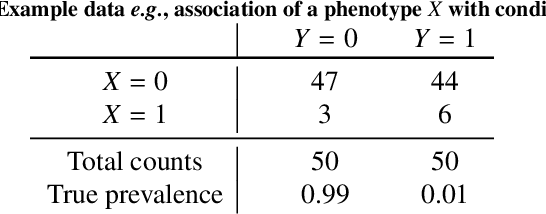
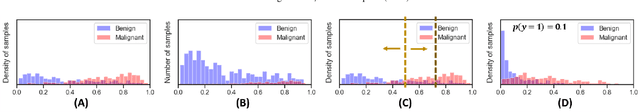
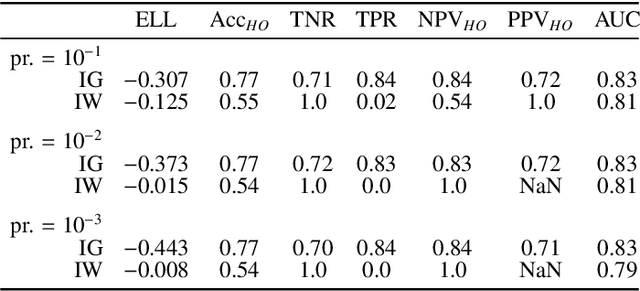
Datasets are rarely a realistic approximation of the target population. Say, prevalence is misrepresented, image quality is above clinical standards, etc. This mismatch is known as sampling bias. Sampling biases are a major hindrance for machine learning models. They cause significant gaps between model performance in the lab and in the real world. Our work is a solution to prevalence bias. Prevalence bias is the discrepancy between the prevalence of a pathology and its sampling rate in the training dataset, introduced upon collecting data or due to the practioner rebalancing the training batches. This paper lays the theoretical and computational framework for training models, and for prediction, in the presence of prevalence bias. Concretely a bias-corrected loss function, as well as bias-corrected predictive rules, are derived under the principles of Bayesian risk minimization. The loss exhibits a direct connection to the information gain. It offers a principled alternative to heuristic training losses and complements test-time procedures based on selecting an operating point from summary curves. It integrates seamlessly in the current paradigm of (deep) learning using stochastic backpropagation and naturally with Bayesian models.