 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Multicenter Benchmark of Multiple Instance Learning Models for Lymphoma Subtyping from HE-stained Whole Slide Images
Dec 16, 2025Timely and accurate lymphoma diagnosis is essential for guiding cancer treatment. Standard diagnostic practice combines hematoxylin and eosin (HE)-stained whole slide images with immunohistochemistry, flow cytometry, and molecular genetic tests to determine lymphoma subtypes, a process requiring costly equipment, skilled personnel, and causing treatment delays. Deep learning methods could assist pathologists by extracting diagnostic information from routinely available HE-stained slides, yet comprehensive benchmarks for lymphoma subtyping on multicenter data are lacking. In this work, we present the first multicenter lymphoma benchmarking dataset covering four common lymphoma subtypes and healthy control tissue. We systematically evaluate five publicly available pathology foundation models (H-optimus-1, H0-mini, Virchow2, UNI2, Titan) combined with attention-based (AB-MIL) and transformer-based (TransMIL) multiple instance learning aggregators across three magnifications (10x, 20x, 40x). On in-distribution test sets, models achieve multiclass balanced accuracies exceeding 80% across all magnifications, with all foundation models performing similarly and both aggregation methods showing comparable results. The magnification study reveals that 40x resolution is sufficient, with no performance gains from higher resolutions or cross-magnification aggregation. However, on out-of-distribution test sets, performance drops substantially to around 60%, highlighting significant generalization challenges. To advance the field, larger multicenter studies covering additional rare lymphoma subtypes are needed. We provide an automated benchmarking pipeline to facilitate such future research.
Neural Cellular Automata for Weakly Supervised Segmentation of White Blood Cells
Aug 17, 2025The detection and segmentation of white blood cells in blood smear images is a key step in medical diagnostics, supporting various downstream tasks such as automated blood cell counting, morphological analysis, cell classification, and disease diagnosis and monitoring. Training robust and accurate models requires large amounts of labeled data, which is both time-consuming and expensive to acquire. In this work, we propose a novel approach for weakly supervised segmentation using neural cellular automata (NCA-WSS). By leveraging the feature maps generated by NCA during classification, we can extract segmentation masks without the need for retraining with segmentation labels. We evaluate our method on three white blood cell microscopy datasets and demonstrate that NCA-WSS significantly outperforms existing weakly supervised approaches. Our work illustrates the potential of NCA for both classification and segmentation in a weakly supervised framework, providing a scalable and efficient solution for medical image analysis.
Attention Pooling Enhances NCA-based Classification of Microscopy Images
Aug 17, 2025Neural Cellular Automata (NCA) offer a robust and interpretable approach to image classification, making them a promising choice for microscopy image analysis. However, a performance gap remains between NCA and larger, more complex architectures. We address this challenge by integrating attention pooling with NCA to enhance feature extraction and improve classification accuracy. The attention pooling mechanism refines the focus on the most informative regions, leading to more accurate predictions. We evaluate our method on eight diverse microscopy image datasets and demonstrate that our approach significantly outperforms existing NCA methods while remaining parameter-efficient and explainable. Furthermore, we compare our method with traditional lightweight convolutional neural network and vision transformer architectures, showing improved performance while maintaining a significantly lower parameter count. Our results highlight the potential of NCA-based models an alternative for explainable image classification.
RedDino: A foundation model for red blood cell analysis
Aug 11, 2025Red blood cells (RBCs) are essential to human health, and their precise morphological analysis is important for diagnosing hematological disorders. Despite the promise of foundation models in medical diagnostics, comprehensive AI solutions for RBC analysis remain scarce. We present RedDino, a self-supervised foundation model designed for RBC image analysis. RedDino uses an RBC-specific adaptation of the DINOv2 self-supervised learning framework and is trained on a curated dataset of 1.25 million RBC images from diverse acquisition modalities and sources. Extensive evaluations show that RedDino outperforms existing state-of-the-art models on RBC shape classification. Through assessments including linear probing and nearest neighbor classification, we confirm its strong feature representations and generalization ability. Our main contributions are: (1) a foundation model tailored for RBC analysis, (2) ablation studies exploring DINOv2 configurations for RBC modeling, and (3) a detailed evaluation of generalization performance. RedDino addresses key challenges in computational hematology by capturing nuanced morphological features, advancing the development of reliable diagnostic tools. The source code and pretrained models for RedDino are available at https://github.com/Snarci/RedDino, and the pretrained models can be downloaded from our Hugging Face collection at https://huggingface.co/collections/Snarcy/reddino-689a13e29241d2e5690202fc
CytoSAE: Interpretable Cell Embeddings for Hematology
Jul 16, 2025



Sparse autoencoders (SAEs) emerged as a promising tool for mechanistic interpretability of transformer-based foundation models. Very recently, SAEs were also adopted for the visual domain, enabling the discovery of visual concepts and their patch-wise attribution to tokens in the transformer model. While a growing number of foundation models emerged for medical imaging, tools for explaining their inferences are still lacking. In this work, we show the applicability of SAEs for hematology. We propose CytoSAE, a sparse autoencoder which is trained on over 40,000 peripheral blood single-cell images. CytoSAE generalizes to diverse and out-of-domain datasets, including bone marrow cytology, where it identifies morphologically relevant concepts which we validated with medical experts. Furthermore, we demonstrate scenarios in which CytoSAE can generate patient-specific and disease-specific concepts, enabling the detection of pathognomonic cells and localized cellular abnormalities at the patch level. We quantified the effect of concepts on a patient-level AML subtype classification task and show that CytoSAE concepts reach performance comparable to the state-of-the-art, while offering explainability on the sub-cellular level. Source code and model weights are available at https://github.com/dynamical-inference/cytosae.
Addressing pitfalls in implicit unobserved confounding synthesis using explicit block hierarchical ancestral sampling
Mar 12, 2025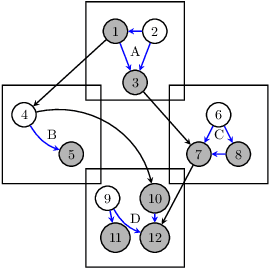
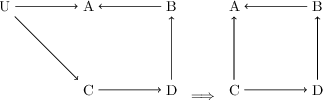
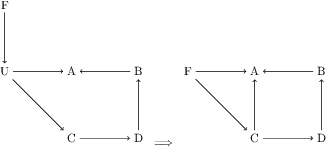
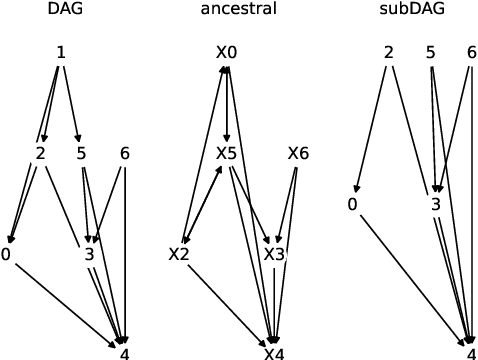
Unbiased data synthesis is crucial for evaluating causal discovery algorithms in the presence of unobserved confounding, given the scarcity of real-world datasets. A common approach, implicit parameterization, encodes unobserved confounding by modifying the off-diagonal entries of the idiosyncratic covariance matrix while preserving positive definiteness. Within this approach, state-of-the-art protocols have two distinct issues that hinder unbiased sampling from the complete space of causal models: first, the use of diagonally dominant constructions, which restrict the spectrum of partial correlation matrices; and second, the restriction of possible graphical structures when sampling bidirected edges, unnecessarily ruling out valid causal models. To address these limitations, we propose an improved explicit modeling approach for unobserved confounding, leveraging block-hierarchical ancestral generation of ground truth causal graphs. Algorithms for converting the ground truth DAG into ancestral graph is provided so that the output of causal discovery algorithms could be compared with. We prove that our approach fully covers the space of causal models, including those generated by the implicit parameterization, thus enabling more robust evaluation of methods for causal discovery and inference.
CellPilot: A unified approach to automatic and interactive segmentation in histopathology
Nov 23, 2024



Histopathology, the microscopic study of diseased tissue, is increasingly digitized, enabling improved visualization and streamlined workflows. An important task in histopathology is the segmentation of cells and glands, essential for determining shape and frequencies that can serve as indicators of disease. Deep learning tools are widely used in histopathology. However, variability in tissue appearance and cell morphology presents challenges for achieving reliable segmentation, often requiring manual correction to improve accuracy. This work introduces CellPilot, a framework that bridges the gap between automatic and interactive segmentation by providing initial automatic segmentation as well as guided interactive refinement. Our model was trained on over 675,000 masks of nine diverse cell and gland segmentation datasets, spanning 16 organs. CellPilot demonstrates superior performance compared to other interactive tools on three held-out histopathological datasets while enabling automatic segmentation. We make the model and a graphical user interface designed to assist practitioners in creating large-scale annotated datasets available as open-source, fostering the development of more robust and generalized diagnostic models.
CellPilot
Nov 23, 2024Histopathology, the microscopic study of diseased tissue, is increasingly digitized, enabling improved visualization and streamlined workflows. An important task in histopathology is the segmentation of cells and glands, essential for determining shape and frequencies that can serve as indicators of disease. Deep learning tools are widely used in histopathology. However, variability in tissue appearance and cell morphology presents challenges for achieving reliable segmentation, often requiring manual correction to improve accuracy. This work introduces CellPilot, a framework that bridges the gap between automatic and interactive segmentation by providing initial automatic segmentation as well as guided interactive refinement. Our model was trained on over 675,000 masks of nine diverse cell and gland segmentation datasets, spanning 16 organs. CellPilot demonstrates superior performance compared to other interactive tools on three held-out histopathological datasets while enabling automatic segmentation. We make the model and a graphical user interface designed to assist practitioners in creating large-scale annotated datasets available as open-source, fostering the development of more robust and generalized diagnostic models.
UNICORN: A Deep Learning Model for Integrating Multi-Stain Data in Histopathology
Sep 26, 2024Background: The integration of multi-stain histopathology images through deep learning poses a significant challenge in digital histopathology. Current multi-modal approaches struggle with data heterogeneity and missing data. This study aims to overcome these limitations by developing a novel transformer model for multi-stain integration that can handle missing data during training as well as inference. Methods: We propose UNICORN (UNiversal modality Integration Network for CORonary classificatioN) a multi-modal transformer capable of processing multi-stain histopathology for atherosclerosis severity class prediction. The architecture comprises a two-stage, end-to-end trainable model with specialized modules utilizing transformer self-attention blocks. The initial stage employs domain-specific expert modules to extract features from each modality. In the subsequent stage, an aggregation expert module integrates these features by learning the interactions between the different data modalities. Results: Evaluation was performed using a multi-class dataset of atherosclerotic lesions from the Munich Cardiovascular Studies Biobank (MISSION), using over 4,000 paired multi-stain whole slide images (WSIs) from 170 deceased individuals on 7 prespecified segments of the coronary tree, each stained according to four histopathological protocols. UNICORN achieved a classification accuracy of 0.67, outperforming other state-of-the-art models. The model effectively identifies relevant tissue phenotypes across stainings and implicitly models disease progression. Conclusion: Our proposed multi-modal transformer model addresses key challenges in medical data analysis, including data heterogeneity and missing modalities. Explainability and the model's effectiveness in predicting atherosclerosis progression underscores its potential for broader applications in medical research.
Multimodal Analysis of White Blood Cell Differentiation in Acute Myeloid Leukemia Patients using a β-Variational Autoencoder
Aug 13, 2024Biomedical imaging and RNA sequencing with single-cell resolution improves our understanding of white blood cell diseases like leukemia. By combining morphological and transcriptomic data, we can gain insights into cellular functions and trajectoriess involved in blood cell differentiation. However, existing methodologies struggle with integrating morphological and transcriptomic data, leaving a significant research gap in comprehensively understanding the dynamics of cell differentiation. Here, we introduce an unsupervised method that explores and reconstructs these two modalities and uncovers the relationship between different subtypes of white blood cells from human peripheral blood smears in terms of morphology and their corresponding transcriptome. Our method is based on a beta-variational autoencoder (\beta-VAE) with a customized loss function, incorporating a R-CNN architecture to distinguish single-cell from background and to minimize any interference from artifacts. This implementation of \beta-VAE shows good reconstruction capability along with continuous latent embeddings, while maintaining clear differentiation between single-cell classes. Our novel approach is especially helpful to uncover the correlation of two latent features in complex biological processes such as formation of granules in the cell (granulopoiesis) with gene expression patterns. It thus provides a unique tool to improve the understanding of white blood cell maturation for biomedicine and diagnostics.