 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTowards Spatial Transcriptomics-driven Pathology Foundation Models
Feb 15, 2026Spatial transcriptomics (ST) provides spatially resolved measurements of gene expression, enabling characterization of the molecular landscape of human tissue beyond histological assessment as well as localized readouts that can be aligned with morphology. Concurrently, the success of multimodal foundation models that integrate vision with complementary modalities suggests that morphomolecular coupling between local expression and morphology can be systematically used to improve histological representations themselves. We introduce Spatial Expression-Aligned Learning (SEAL), a vision-omics self-supervised learning framework that infuses localized molecular information into pathology vision encoders. Rather than training new encoders from scratch, SEAL is designed as a parameter-efficient vision-omics finetuning method that can be flexibly applied to widely used pathology foundation models. We instantiate SEAL by training on over 700,000 paired gene expression spot-tissue region examples spanning tumor and normal samples from 14 organs. Tested across 38 slide-level and 15 patch-level downstream tasks, SEAL provides a drop-in replacement for pathology foundation models that consistently improves performance over widely used vision-only and ST prediction baselines on slide-level molecular status, pathway activity, and treatment response prediction, as well as patch-level gene expression prediction tasks. Additionally, SEAL encoders exhibit robust domain generalization on out-of-distribution evaluations and enable new cross-modal capabilities such as gene-to-image retrieval. Our work proposes a general framework for ST-guided finetuning of pathology foundation models, showing that augmenting existing models with localized molecular supervision is an effective and practical step for improving visual representations and expanding their cross-modal utility.
AI-driven 3D Spatial Transcriptomics
Feb 25, 2025



A comprehensive three-dimensional (3D) map of tissue architecture and gene expression is crucial for illuminating the complexity and heterogeneity of tissues across diverse biomedical applications. However, most spatial transcriptomics (ST) approaches remain limited to two-dimensional (2D) sections of tissue. Although current 3D ST methods hold promise, they typically require extensive tissue sectioning, are complex, are not compatible with non-destructive 3D tissue imaging technologies, and often lack scalability. Here, we present VOlumetrically Resolved Transcriptomics EXpression (VORTEX), an AI framework that leverages 3D tissue morphology and minimal 2D ST to predict volumetric 3D ST. By pretraining on diverse 3D morphology-transcriptomic pairs from heterogeneous tissue samples and then fine-tuning on minimal 2D ST data from a specific volume of interest, VORTEX learns both generic tissue-related and sample-specific morphological correlates of gene expression. This approach enables dense, high-throughput, and fast 3D ST, scaling seamlessly to large tissue volumes far beyond the reach of existing 3D ST techniques. By offering a cost-effective and minimally destructive route to obtaining volumetric molecular insights, we anticipate that VORTEX will accelerate biomarker discovery and our understanding of morphomolecular associations and cell states in complex tissues. Interactive 3D ST volumes can be viewed at https://vortex-demo.github.io/
HEST-1k: A Dataset for Spatial Transcriptomics and Histology Image Analysis
Jun 23, 2024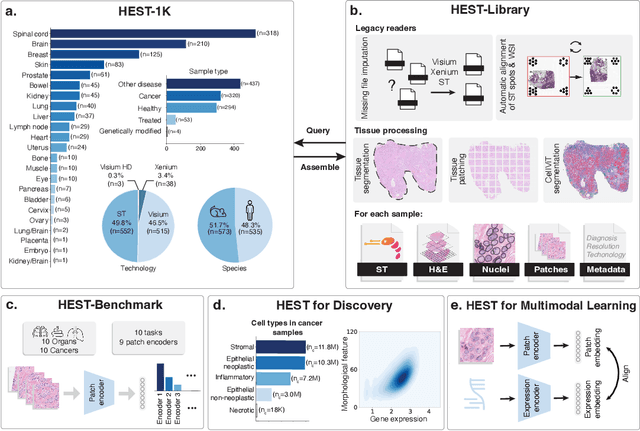
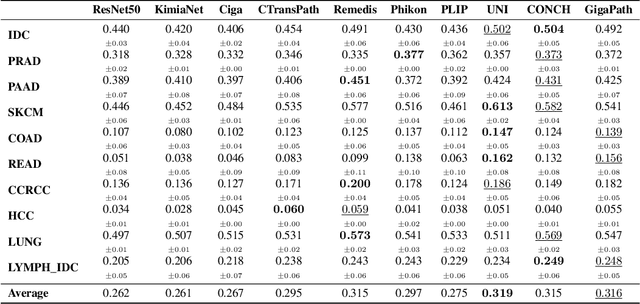
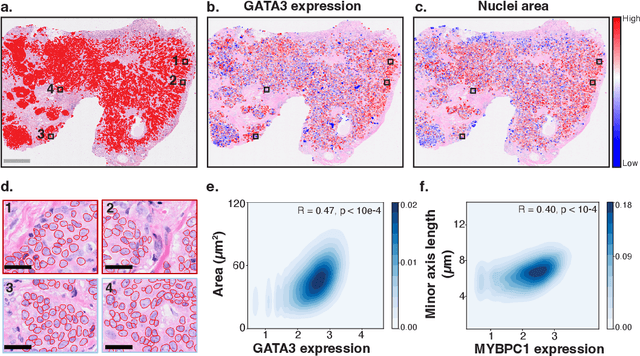
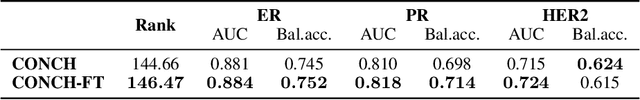
Spatial transcriptomics (ST) enables interrogating the molecular composition of tissue with ever-increasing resolution, depth, and sensitivity. However, costs, rapidly evolving technology, and lack of standards have constrained computational methods in ST to narrow tasks and small cohorts. In addition, the underlying tissue morphology as reflected by H&E-stained whole slide images (WSIs) encodes rich information often overlooked in ST studies. Here, we introduce HEST-1k, a collection of 1,108 spatial transcriptomic profiles, each linked to a WSI and metadata. HEST-1k was assembled using HEST-Library from 131 public and internal cohorts encompassing 25 organs, two species (Homo Sapiens and Mus Musculus), and 320 cancer samples from 25 cancer types. HEST-1k processing enabled the identification of 1.5 million expression--morphology pairs and 60 million nuclei. HEST-1k is tested on three use cases: (1) benchmarking foundation models for histopathology (HEST-Benchmark), (2) biomarker identification, and (3) multimodal representation learning. HEST-1k, HEST-Library, and HEST-Benchmark can be freely accessed via https://github.com/mahmoodlab/hest.