 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeNOVA: An Agentic Framework for Automated Histopathology Analysis and Discovery
Nov 14, 2025



Digitized histopathology analysis involves complex, time-intensive workflows and specialized expertise, limiting its accessibility. We introduce NOVA, an agentic framework that translates scientific queries into executable analysis pipelines by iteratively generating and running Python code. NOVA integrates 49 domain-specific tools (e.g., nuclei segmentation, whole-slide encoding) built on open-source software, and can also create new tools ad hoc. To evaluate such systems, we present SlideQuest, a 90-question benchmark -- verified by pathologists and biomedical scientists -- spanning data processing, quantitative analysis, and hypothesis testing. Unlike prior biomedical benchmarks focused on knowledge recall or diagnostic QA, SlideQuest demands multi-step reasoning, iterative coding, and computational problem solving. Quantitative evaluation shows NOVA outperforms coding-agent baselines, and a pathologist-verified case study links morphology to prognostically relevant PAM50 subtypes, demonstrating its scalable discovery potential.
Multimodal Whole Slide Foundation Model for Pathology
Nov 29, 2024



The field of computational pathology has been transformed with recent advances in foundation models that encode histopathology region-of-interests (ROIs) into versatile and transferable feature representations via self-supervised learning (SSL). However, translating these advancements to address complex clinical challenges at the patient and slide level remains constrained by limited clinical data in disease-specific cohorts, especially for rare clinical conditions. We propose TITAN, a multimodal whole slide foundation model pretrained using 335,645 WSIs via visual self-supervised learning and vision-language alignment with corresponding pathology reports and 423,122 synthetic captions generated from a multimodal generative AI copilot for pathology. Without any finetuning or requiring clinical labels, TITAN can extract general-purpose slide representations and generate pathology reports that generalize to resource-limited clinical scenarios such as rare disease retrieval and cancer prognosis. We evaluate TITAN on diverse clinical tasks and find that TITAN outperforms both ROI and slide foundation models across machine learning settings such as linear probing, few-shot and zero-shot classification, rare cancer retrieval and cross-modal retrieval, and pathology report generation.
Multimodal Prototyping for cancer survival prediction
Jun 28, 2024



Multimodal survival methods combining gigapixel histology whole-slide images (WSIs) and transcriptomic profiles are particularly promising for patient prognostication and stratification. Current approaches involve tokenizing the WSIs into smaller patches (>10,000 patches) and transcriptomics into gene groups, which are then integrated using a Transformer for predicting outcomes. However, this process generates many tokens, which leads to high memory requirements for computing attention and complicates post-hoc interpretability analyses. Instead, we hypothesize that we can: (1) effectively summarize the morphological content of a WSI by condensing its constituting tokens using morphological prototypes, achieving more than 300x compression; and (2) accurately characterize cellular functions by encoding the transcriptomic profile with biological pathway prototypes, all in an unsupervised fashion. The resulting multimodal tokens are then processed by a fusion network, either with a Transformer or an optimal transport cross-alignment, which now operates with a small and fixed number of tokens without approximations. Extensive evaluation on six cancer types shows that our framework outperforms state-of-the-art methods with much less computation while unlocking new interpretability analyses.
HEST-1k: A Dataset for Spatial Transcriptomics and Histology Image Analysis
Jun 23, 2024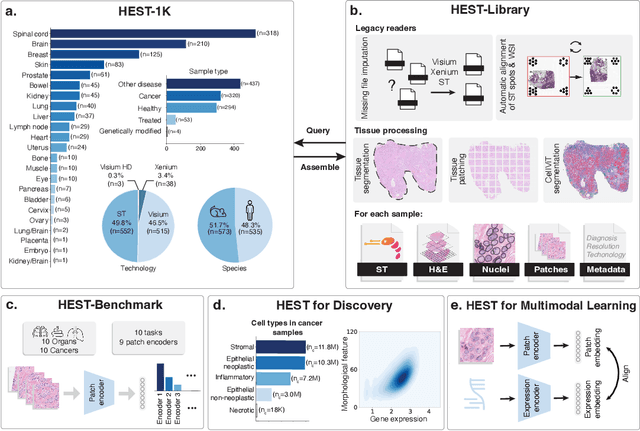
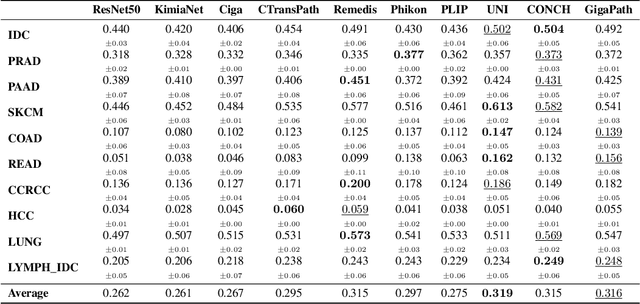
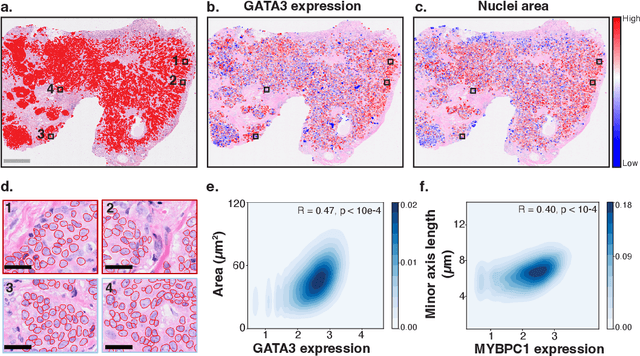
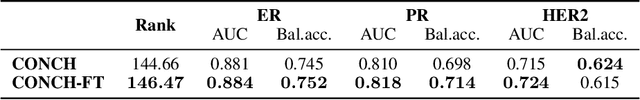
Spatial transcriptomics (ST) enables interrogating the molecular composition of tissue with ever-increasing resolution, depth, and sensitivity. However, costs, rapidly evolving technology, and lack of standards have constrained computational methods in ST to narrow tasks and small cohorts. In addition, the underlying tissue morphology as reflected by H&E-stained whole slide images (WSIs) encodes rich information often overlooked in ST studies. Here, we introduce HEST-1k, a collection of 1,108 spatial transcriptomic profiles, each linked to a WSI and metadata. HEST-1k was assembled using HEST-Library from 131 public and internal cohorts encompassing 25 organs, two species (Homo Sapiens and Mus Musculus), and 320 cancer samples from 25 cancer types. HEST-1k processing enabled the identification of 1.5 million expression--morphology pairs and 60 million nuclei. HEST-1k is tested on three use cases: (1) benchmarking foundation models for histopathology (HEST-Benchmark), (2) biomarker identification, and (3) multimodal representation learning. HEST-1k, HEST-Library, and HEST-Benchmark can be freely accessed via https://github.com/mahmoodlab/hest.