 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDALPHIN: Benchmarking Digital Pathology AI Copilots Against Pathologists on an Open Multicentric Dataset
May 05, 2026Foundation models with visual question answering capabilities for digital pathology are emerging. Such unprecedented technology requires independent benchmarking to assess its potential in assisting pathologists in routine diagnostics. We created DALPHIN, the first multicentric open benchmark for pathology AI copilots, comprising 1236 images from 300 cases, spanning 130 rare to common diagnoses, 6 countries, and 14 subspecialties. The DALPHIN design and dataset are introduced alongside a human performance benchmark of 31 pathologists from 10 countries with varying expertise. We report results for two general-purpose (GPT-5, Gemini 2.5 Pro) and one pathology-specific copilot (PathChat+) for sequential and independent answer generation. We observed no statistically significant difference from expert-level performance in four of six tasks for PathChat, 2/6 tasks for Gemini, and 1/6 tasks for GPT. DALPHIN is publicly released with sequestered, indirectly accessible ground truth to foster robust and enduring benchmarking. Data, methods, and the evaluation platform are accessible through dalphin.grand-challenge.org.
A multimodal and temporal foundation model for virtual patient representations at healthcare system scale
Apr 21, 2026Modern medicine generates vast multimodal data across siloed systems, yet no existing model integrates the full breadth and temporal depth of the clinical record into a unified patient representation. We introduce Apollo, a multimodal temporal foundation model trained and evaluated on over three decades of longitudinal hospital records from a major US hospital system, composed of 25 billion records from 7.2 million patients, representing 28 distinct medical modalities and 12 major medical specialties. Apollo learns a unified representation space integrating over 100 thousand unique medical events in our clinical vocabulary as well as images and clinical text. This "atlas of medical concepts" forms a computational substrate for modeling entire patient care journeys comprised of sequences of structured and unstructured events, which are compressed by Apollo into virtual patient representations. To assess the potential of these whole-patient representations, we created 322 prognosis and retrieval tasks from a held-out test set of 1.4 million patients. We demonstrate the generalized clinical forecasting potential of Apollo embeddings, including predicting new disease onset risk up to five years in advance (95 tasks), disease progression (78 tasks), treatment response (59 tasks), risk of treatment-related adverse events (17 tasks), and hospital operations endpoints (12 tasks). Using feature attribution techniques, we show that model predictions align with clinically-interpretable multimodal biomarkers. We evaluate semantic similarity search on 61 retrieval tasks, and moreover demonstrate the potential of Apollo as a multimodal medical search engine using text and image queries. Together, these modeling capabilities establish the foundation for computable medicine, where the full context of patient care becomes accessible to computational reasoning.
Mixture of Mini Experts: Overcoming the Linear Layer Bottleneck in Multiple Instance Learning
Mar 23, 2026Multiple Instance Learning (MIL) is the predominant framework for classifying gigapixel whole-slide images in computational pathology. MIL follows a sequence of 1) extracting patch features, 2) applying a linear layer to obtain task-specific patch features, and 3) aggregating the patches into a slide feature for classification. While substantial efforts have been devoted to optimizing patch feature extraction and aggregation, none have yet addressed the second point, the critical layer which transforms general-purpose features into task-specific features. We hypothesize that this layer constitutes an overlooked performance bottleneck and that stronger representations can be achieved with a low-rank transformation tailored to each patch's phenotype, yielding synergistic effects with any of the existing MIL approaches. To this end, we introduce MAMMOTH, a parameter-efficient, multi-head mixture of experts module designed to improve the performance of any MIL model with minimal alterations to the total number of parameters. Across eight MIL methods and 19 different classification tasks, we find that such task-specific transformation has a larger effect on performance than the choice of aggregation method. For instance, when equipped with MAMMOTH, even simple methods such as max or mean pooling attain higher average performance than any method with the standard linear layer. Overall, MAMMOTH improves performance in 130 of the 152 examined configurations, with an average $+3.8\%$ change in performance. Code is available at https://github.com/mahmoodlab/mammoth.
Towards Spatial Transcriptomics-driven Pathology Foundation Models
Feb 15, 2026Spatial transcriptomics (ST) provides spatially resolved measurements of gene expression, enabling characterization of the molecular landscape of human tissue beyond histological assessment as well as localized readouts that can be aligned with morphology. Concurrently, the success of multimodal foundation models that integrate vision with complementary modalities suggests that morphomolecular coupling between local expression and morphology can be systematically used to improve histological representations themselves. We introduce Spatial Expression-Aligned Learning (SEAL), a vision-omics self-supervised learning framework that infuses localized molecular information into pathology vision encoders. Rather than training new encoders from scratch, SEAL is designed as a parameter-efficient vision-omics finetuning method that can be flexibly applied to widely used pathology foundation models. We instantiate SEAL by training on over 700,000 paired gene expression spot-tissue region examples spanning tumor and normal samples from 14 organs. Tested across 38 slide-level and 15 patch-level downstream tasks, SEAL provides a drop-in replacement for pathology foundation models that consistently improves performance over widely used vision-only and ST prediction baselines on slide-level molecular status, pathway activity, and treatment response prediction, as well as patch-level gene expression prediction tasks. Additionally, SEAL encoders exhibit robust domain generalization on out-of-distribution evaluations and enable new cross-modal capabilities such as gene-to-image retrieval. Our work proposes a general framework for ST-guided finetuning of pathology foundation models, showing that augmenting existing models with localized molecular supervision is an effective and practical step for improving visual representations and expanding their cross-modal utility.
NOVA: An Agentic Framework for Automated Histopathology Analysis and Discovery
Nov 14, 2025



Digitized histopathology analysis involves complex, time-intensive workflows and specialized expertise, limiting its accessibility. We introduce NOVA, an agentic framework that translates scientific queries into executable analysis pipelines by iteratively generating and running Python code. NOVA integrates 49 domain-specific tools (e.g., nuclei segmentation, whole-slide encoding) built on open-source software, and can also create new tools ad hoc. To evaluate such systems, we present SlideQuest, a 90-question benchmark -- verified by pathologists and biomedical scientists -- spanning data processing, quantitative analysis, and hypothesis testing. Unlike prior biomedical benchmarks focused on knowledge recall or diagnostic QA, SlideQuest demands multi-step reasoning, iterative coding, and computational problem solving. Quantitative evaluation shows NOVA outperforms coding-agent baselines, and a pathologist-verified case study links morphology to prognostically relevant PAM50 subtypes, demonstrating its scalable discovery potential.
Evidence-based diagnostic reasoning with multi-agent copilot for human pathology
Jun 26, 2025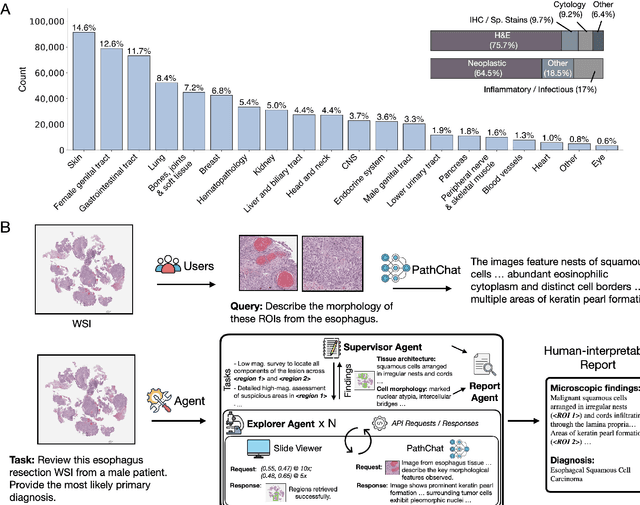
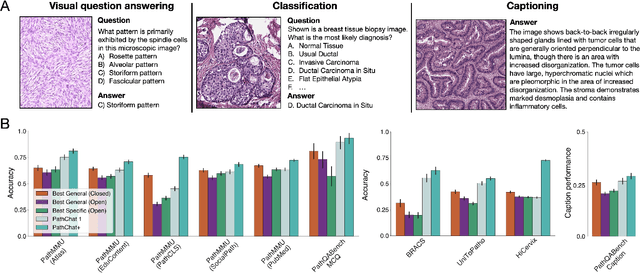
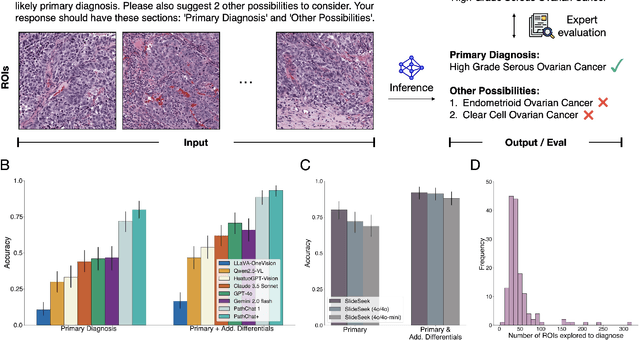
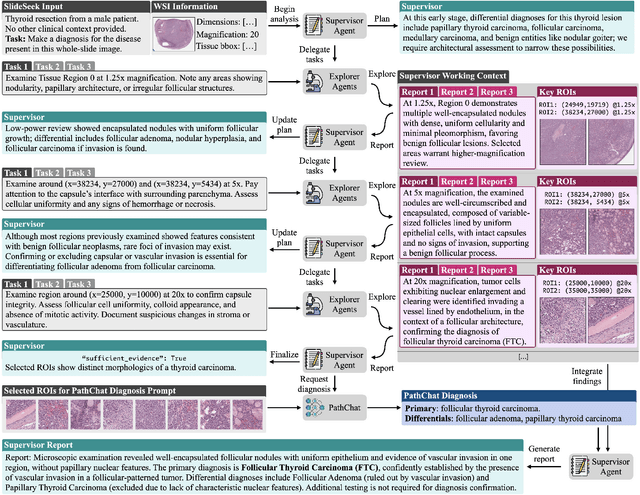
Pathology is experiencing rapid digital transformation driven by whole-slide imaging and artificial intelligence (AI). While deep learning-based computational pathology has achieved notable success, traditional models primarily focus on image analysis without integrating natural language instruction or rich, text-based context. Current multimodal large language models (MLLMs) in computational pathology face limitations, including insufficient training data, inadequate support and evaluation for multi-image understanding, and a lack of autonomous, diagnostic reasoning capabilities. To address these limitations, we introduce PathChat+, a new MLLM specifically designed for human pathology, trained on over 1 million diverse, pathology-specific instruction samples and nearly 5.5 million question answer turns. Extensive evaluations across diverse pathology benchmarks demonstrated that PathChat+ substantially outperforms the prior PathChat copilot, as well as both state-of-the-art (SOTA) general-purpose and other pathology-specific models. Furthermore, we present SlideSeek, a reasoning-enabled multi-agent AI system leveraging PathChat+ to autonomously evaluate gigapixel whole-slide images (WSIs) through iterative, hierarchical diagnostic reasoning, reaching high accuracy on DDxBench, a challenging open-ended differential diagnosis benchmark, while also capable of generating visually grounded, humanly-interpretable summary reports.
Do Multiple Instance Learning Models Transfer?
Jun 11, 2025Multiple Instance Learning (MIL) is a cornerstone approach in computational pathology (CPath) for generating clinically meaningful slide-level embeddings from gigapixel tissue images. However, MIL often struggles with small, weakly supervised clinical datasets. In contrast to fields such as NLP and conventional computer vision, where transfer learning is widely used to address data scarcity, the transferability of MIL models remains poorly understood. In this study, we systematically evaluate the transfer learning capabilities of pretrained MIL models by assessing 11 models across 21 pretraining tasks for morphological and molecular subtype prediction. Our results show that pretrained MIL models, even when trained on different organs than the target task, consistently outperform models trained from scratch. Moreover, pretraining on pancancer datasets enables strong generalization across organs and tasks, outperforming slide foundation models while using substantially less pretraining data. These findings highlight the robust adaptability of MIL models and demonstrate the benefits of leveraging transfer learning to boost performance in CPath. Lastly, we provide a resource which standardizes the implementation of MIL models and collection of pretrained model weights on popular CPath tasks, available at https://github.com/mahmoodlab/MIL-Lab
AI-driven 3D Spatial Transcriptomics
Feb 25, 2025



A comprehensive three-dimensional (3D) map of tissue architecture and gene expression is crucial for illuminating the complexity and heterogeneity of tissues across diverse biomedical applications. However, most spatial transcriptomics (ST) approaches remain limited to two-dimensional (2D) sections of tissue. Although current 3D ST methods hold promise, they typically require extensive tissue sectioning, are complex, are not compatible with non-destructive 3D tissue imaging technologies, and often lack scalability. Here, we present VOlumetrically Resolved Transcriptomics EXpression (VORTEX), an AI framework that leverages 3D tissue morphology and minimal 2D ST to predict volumetric 3D ST. By pretraining on diverse 3D morphology-transcriptomic pairs from heterogeneous tissue samples and then fine-tuning on minimal 2D ST data from a specific volume of interest, VORTEX learns both generic tissue-related and sample-specific morphological correlates of gene expression. This approach enables dense, high-throughput, and fast 3D ST, scaling seamlessly to large tissue volumes far beyond the reach of existing 3D ST techniques. By offering a cost-effective and minimally destructive route to obtaining volumetric molecular insights, we anticipate that VORTEX will accelerate biomarker discovery and our understanding of morphomolecular associations and cell states in complex tissues. Interactive 3D ST volumes can be viewed at https://vortex-demo.github.io/
Accelerating Data Processing and Benchmarking of AI Models for Pathology
Feb 10, 2025Advances in foundation modeling have reshaped computational pathology. However, the increasing number of available models and lack of standardized benchmarks make it increasingly complex to assess their strengths, limitations, and potential for further development. To address these challenges, we introduce a new suite of software tools for whole-slide image processing, foundation model benchmarking, and curated publicly available tasks. We anticipate that these resources will promote transparency, reproducibility, and continued progress in the field.
Molecular-driven Foundation Model for Oncologic Pathology
Jan 28, 2025Foundation models are reshaping computational pathology by enabling transfer learning, where models pre-trained on vast datasets can be adapted for downstream diagnostic, prognostic, and therapeutic response tasks. Despite these advances, foundation models are still limited in their ability to encode the entire gigapixel whole-slide images without additional training and often lack complementary multimodal data. Here, we introduce Threads, a slide-level foundation model capable of generating universal representations of whole-slide images of any size. Threads was pre-trained using a multimodal learning approach on a diverse cohort of 47,171 hematoxylin and eosin (H&E)-stained tissue sections, paired with corresponding genomic and transcriptomic profiles - the largest such paired dataset to be used for foundation model development to date. This unique training paradigm enables Threads to capture the tissue's underlying molecular composition, yielding powerful representations applicable to a wide array of downstream tasks. In extensive benchmarking across 54 oncology tasks, including clinical subtyping, grading, mutation prediction, immunohistochemistry status determination, treatment response prediction, and survival prediction, Threads outperformed all baselines while demonstrating remarkable generalizability and label efficiency. It is particularly well suited for predicting rare events, further emphasizing its clinical utility. We intend to make the model publicly available for the broader community.