 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMolecular-driven Foundation Model for Oncologic Pathology
Jan 28, 2025Foundation models are reshaping computational pathology by enabling transfer learning, where models pre-trained on vast datasets can be adapted for downstream diagnostic, prognostic, and therapeutic response tasks. Despite these advances, foundation models are still limited in their ability to encode the entire gigapixel whole-slide images without additional training and often lack complementary multimodal data. Here, we introduce Threads, a slide-level foundation model capable of generating universal representations of whole-slide images of any size. Threads was pre-trained using a multimodal learning approach on a diverse cohort of 47,171 hematoxylin and eosin (H&E)-stained tissue sections, paired with corresponding genomic and transcriptomic profiles - the largest such paired dataset to be used for foundation model development to date. This unique training paradigm enables Threads to capture the tissue's underlying molecular composition, yielding powerful representations applicable to a wide array of downstream tasks. In extensive benchmarking across 54 oncology tasks, including clinical subtyping, grading, mutation prediction, immunohistochemistry status determination, treatment response prediction, and survival prediction, Threads outperformed all baselines while demonstrating remarkable generalizability and label efficiency. It is particularly well suited for predicting rare events, further emphasizing its clinical utility. We intend to make the model publicly available for the broader community.
Multimodal Whole Slide Foundation Model for Pathology
Nov 29, 2024



The field of computational pathology has been transformed with recent advances in foundation models that encode histopathology region-of-interests (ROIs) into versatile and transferable feature representations via self-supervised learning (SSL). However, translating these advancements to address complex clinical challenges at the patient and slide level remains constrained by limited clinical data in disease-specific cohorts, especially for rare clinical conditions. We propose TITAN, a multimodal whole slide foundation model pretrained using 335,645 WSIs via visual self-supervised learning and vision-language alignment with corresponding pathology reports and 423,122 synthetic captions generated from a multimodal generative AI copilot for pathology. Without any finetuning or requiring clinical labels, TITAN can extract general-purpose slide representations and generate pathology reports that generalize to resource-limited clinical scenarios such as rare disease retrieval and cancer prognosis. We evaluate TITAN on diverse clinical tasks and find that TITAN outperforms both ROI and slide foundation models across machine learning settings such as linear probing, few-shot and zero-shot classification, rare cancer retrieval and cross-modal retrieval, and pathology report generation.
HEST-1k: A Dataset for Spatial Transcriptomics and Histology Image Analysis
Jun 23, 2024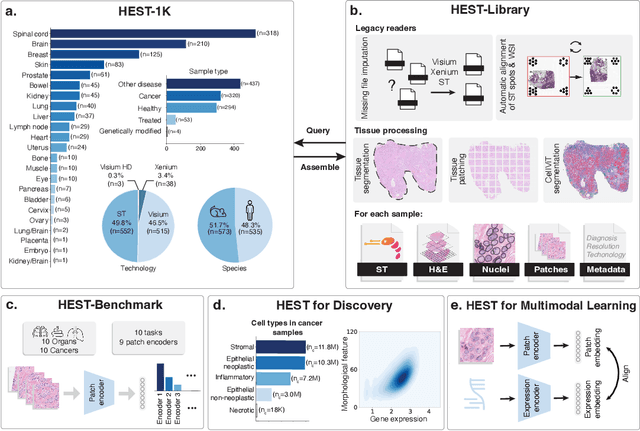
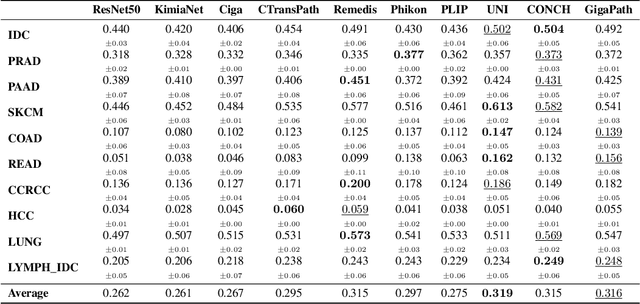
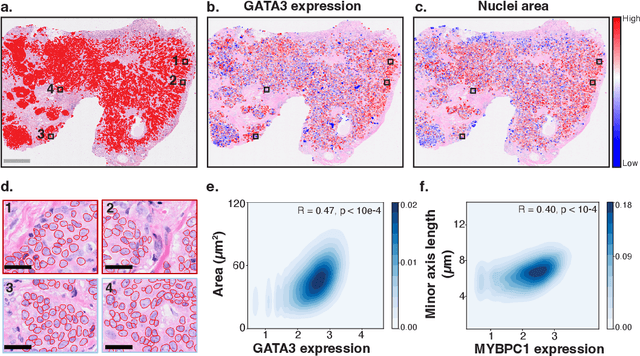
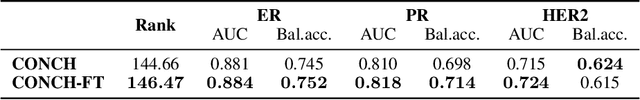
Spatial transcriptomics (ST) enables interrogating the molecular composition of tissue with ever-increasing resolution, depth, and sensitivity. However, costs, rapidly evolving technology, and lack of standards have constrained computational methods in ST to narrow tasks and small cohorts. In addition, the underlying tissue morphology as reflected by H&E-stained whole slide images (WSIs) encodes rich information often overlooked in ST studies. Here, we introduce HEST-1k, a collection of 1,108 spatial transcriptomic profiles, each linked to a WSI and metadata. HEST-1k was assembled using HEST-Library from 131 public and internal cohorts encompassing 25 organs, two species (Homo Sapiens and Mus Musculus), and 320 cancer samples from 25 cancer types. HEST-1k processing enabled the identification of 1.5 million expression--morphology pairs and 60 million nuclei. HEST-1k is tested on three use cases: (1) benchmarking foundation models for histopathology (HEST-Benchmark), (2) biomarker identification, and (3) multimodal representation learning. HEST-1k, HEST-Library, and HEST-Benchmark can be freely accessed via https://github.com/mahmoodlab/hest.
Bridging Diversity and Uncertainty in Active learning with Self-Supervised Pre-Training
Mar 06, 2024



This study addresses the integration of diversity-based and uncertainty-based sampling strategies in active learning, particularly within the context of self-supervised pre-trained models. We introduce a straightforward heuristic called TCM that mitigates the cold start problem while maintaining strong performance across various data levels. By initially applying TypiClust for diversity sampling and subsequently transitioning to uncertainty sampling with Margin, our approach effectively combines the strengths of both strategies. Our experiments demonstrate that TCM consistently outperforms existing methods across various datasets in both low and high data regimes.