 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEvidence-based diagnostic reasoning with multi-agent copilot for human pathology
Jun 26, 2025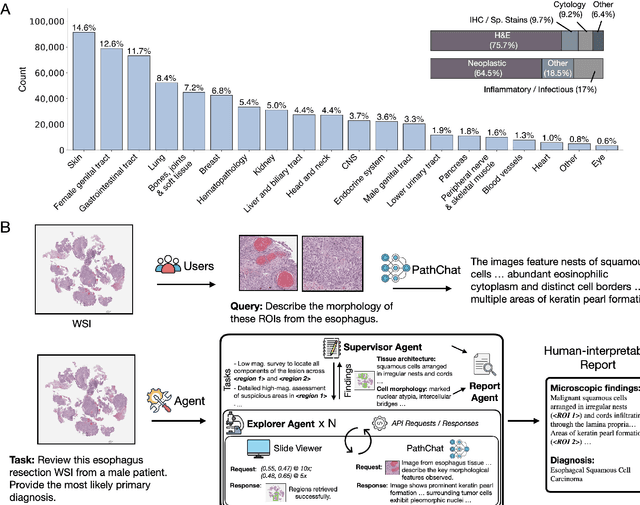
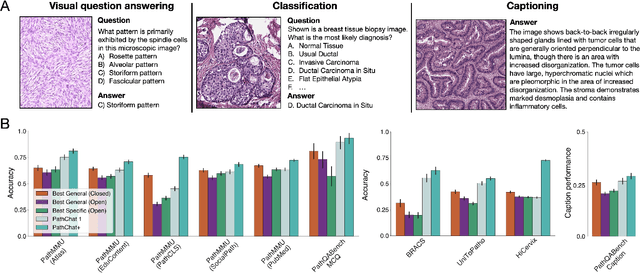
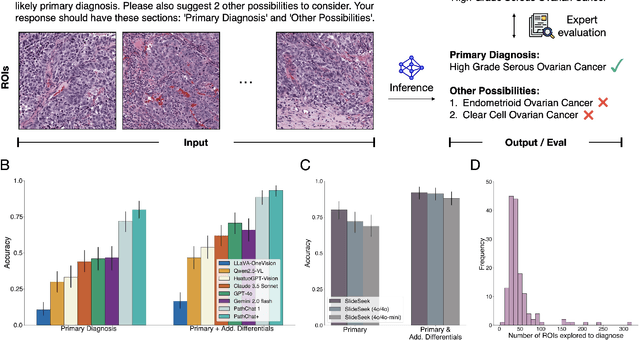
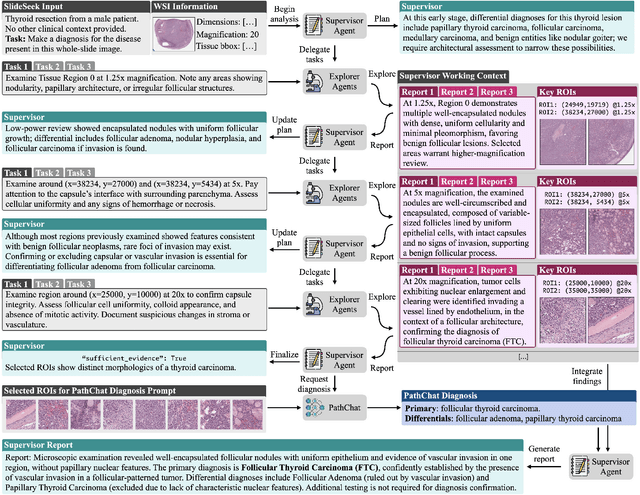
Pathology is experiencing rapid digital transformation driven by whole-slide imaging and artificial intelligence (AI). While deep learning-based computational pathology has achieved notable success, traditional models primarily focus on image analysis without integrating natural language instruction or rich, text-based context. Current multimodal large language models (MLLMs) in computational pathology face limitations, including insufficient training data, inadequate support and evaluation for multi-image understanding, and a lack of autonomous, diagnostic reasoning capabilities. To address these limitations, we introduce PathChat+, a new MLLM specifically designed for human pathology, trained on over 1 million diverse, pathology-specific instruction samples and nearly 5.5 million question answer turns. Extensive evaluations across diverse pathology benchmarks demonstrated that PathChat+ substantially outperforms the prior PathChat copilot, as well as both state-of-the-art (SOTA) general-purpose and other pathology-specific models. Furthermore, we present SlideSeek, a reasoning-enabled multi-agent AI system leveraging PathChat+ to autonomously evaluate gigapixel whole-slide images (WSIs) through iterative, hierarchical diagnostic reasoning, reaching high accuracy on DDxBench, a challenging open-ended differential diagnosis benchmark, while also capable of generating visually grounded, humanly-interpretable summary reports.
Accelerating Data Processing and Benchmarking of AI Models for Pathology
Feb 10, 2025Advances in foundation modeling have reshaped computational pathology. However, the increasing number of available models and lack of standardized benchmarks make it increasingly complex to assess their strengths, limitations, and potential for further development. To address these challenges, we introduce a new suite of software tools for whole-slide image processing, foundation model benchmarking, and curated publicly available tasks. We anticipate that these resources will promote transparency, reproducibility, and continued progress in the field.
Molecular-driven Foundation Model for Oncologic Pathology
Jan 28, 2025Foundation models are reshaping computational pathology by enabling transfer learning, where models pre-trained on vast datasets can be adapted for downstream diagnostic, prognostic, and therapeutic response tasks. Despite these advances, foundation models are still limited in their ability to encode the entire gigapixel whole-slide images without additional training and often lack complementary multimodal data. Here, we introduce Threads, a slide-level foundation model capable of generating universal representations of whole-slide images of any size. Threads was pre-trained using a multimodal learning approach on a diverse cohort of 47,171 hematoxylin and eosin (H&E)-stained tissue sections, paired with corresponding genomic and transcriptomic profiles - the largest such paired dataset to be used for foundation model development to date. This unique training paradigm enables Threads to capture the tissue's underlying molecular composition, yielding powerful representations applicable to a wide array of downstream tasks. In extensive benchmarking across 54 oncology tasks, including clinical subtyping, grading, mutation prediction, immunohistochemistry status determination, treatment response prediction, and survival prediction, Threads outperformed all baselines while demonstrating remarkable generalizability and label efficiency. It is particularly well suited for predicting rare events, further emphasizing its clinical utility. We intend to make the model publicly available for the broader community.
Multistain Pretraining for Slide Representation Learning in Pathology
Aug 05, 2024Developing self-supervised learning (SSL) models that can learn universal and transferable representations of H&E gigapixel whole-slide images (WSIs) is becoming increasingly valuable in computational pathology. These models hold the potential to advance critical tasks such as few-shot classification, slide retrieval, and patient stratification. Existing approaches for slide representation learning extend the principles of SSL from small images (e.g., 224 x 224 patches) to entire slides, usually by aligning two different augmentations (or views) of the slide. Yet the resulting representation remains constrained by the limited clinical and biological diversity of the views. Instead, we postulate that slides stained with multiple markers, such as immunohistochemistry, can be used as different views to form a rich task-agnostic training signal. To this end, we introduce Madeleine, a multimodal pretraining strategy for slide representation learning. Madeleine is trained with a dual global-local cross-stain alignment objective on large cohorts of breast cancer samples (N=4,211 WSIs across five stains) and kidney transplant samples (N=12,070 WSIs across four stains). We demonstrate the quality of slide representations learned by Madeleine on various downstream evaluations, ranging from morphological and molecular classification to prognostic prediction, comprising 21 tasks using 7,299 WSIs from multiple medical centers. Code is available at https://github.com/mahmoodlab/MADELEINE.
Composable Interventions for Language Models
Jul 09, 2024



Test-time interventions for language models can enhance factual accuracy, mitigate harmful outputs, and improve model efficiency without costly retraining. But despite a flood of new methods, different types of interventions are largely developing independently. In practice, multiple interventions must be applied sequentially to the same model, yet we lack standardized ways to study how interventions interact. We fill this gap by introducing composable interventions, a framework to study the effects of using multiple interventions on the same language models, featuring new metrics and a unified codebase. Using our framework, we conduct extensive experiments and compose popular methods from three emerging intervention categories -- Knowledge Editing, Model Compression, and Machine Unlearning. Our results from 310 different compositions uncover meaningful interactions: compression hinders editing and unlearning, composing interventions hinges on their order of application, and popular general-purpose metrics are inadequate for assessing composability. Taken together, our findings showcase clear gaps in composability, suggesting a need for new multi-objective interventions. All of our code is public: https://github.com/hartvigsen-group/composable-interventions.
Transcriptomics-guided Slide Representation Learning in Computational Pathology
May 19, 2024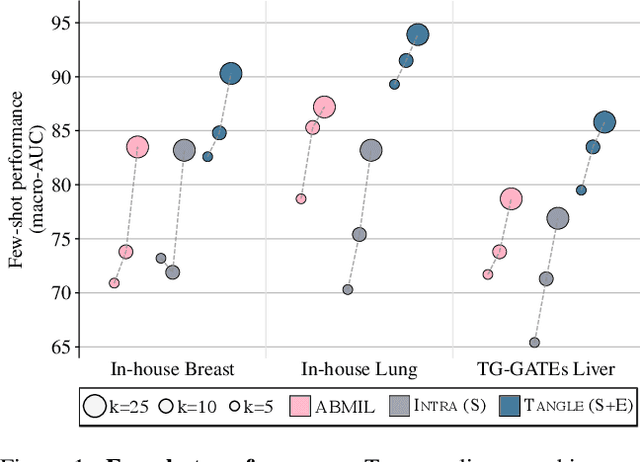
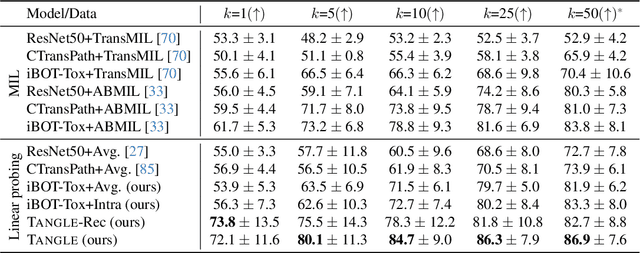
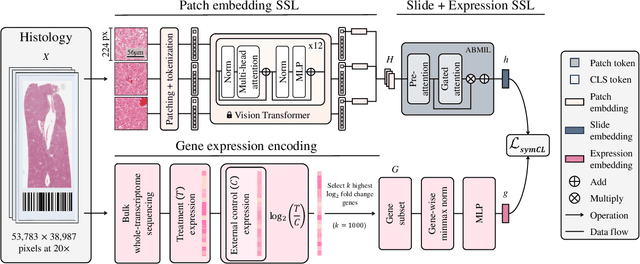
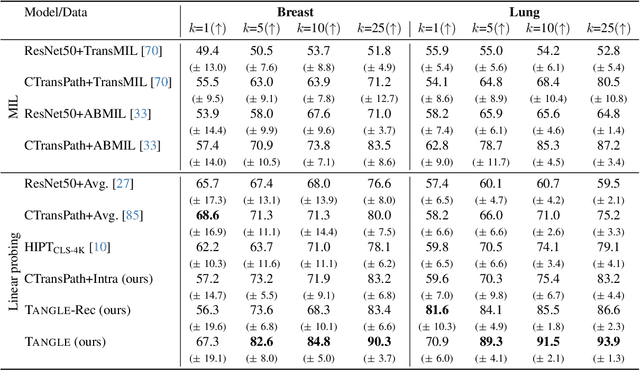
Self-supervised learning (SSL) has been successful in building patch embeddings of small histology images (e.g., 224x224 pixels), but scaling these models to learn slide embeddings from the entirety of giga-pixel whole-slide images (WSIs) remains challenging. Here, we leverage complementary information from gene expression profiles to guide slide representation learning using multimodal pre-training. Expression profiles constitute highly detailed molecular descriptions of a tissue that we hypothesize offer a strong task-agnostic training signal for learning slide embeddings. Our slide and expression (S+E) pre-training strategy, called Tangle, employs modality-specific encoders, the outputs of which are aligned via contrastive learning. Tangle was pre-trained on samples from three different organs: liver (n=6,597 S+E pairs), breast (n=1,020), and lung (n=1,012) from two different species (Homo sapiens and Rattus norvegicus). Across three independent test datasets consisting of 1,265 breast WSIs, 1,946 lung WSIs, and 4,584 liver WSIs, Tangle shows significantly better few-shot performance compared to supervised and SSL baselines. When assessed using prototype-based classification and slide retrieval, Tangle also shows a substantial performance improvement over all baselines. Code available at https://github.com/mahmoodlab/TANGLE.
A General-Purpose Self-Supervised Model for Computational Pathology
Aug 29, 2023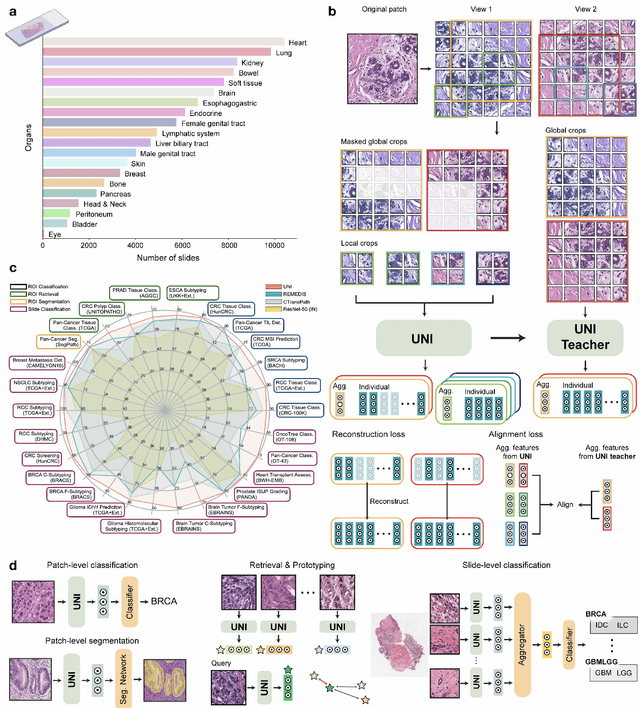

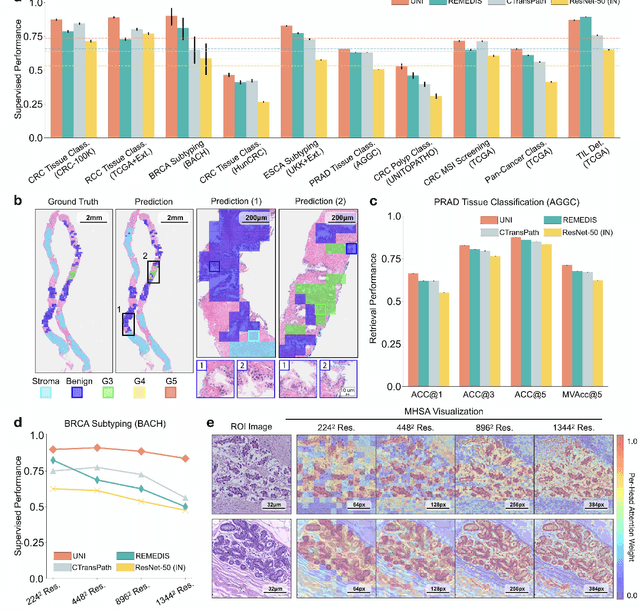

Tissue phenotyping is a fundamental computational pathology (CPath) task in learning objective characterizations of histopathologic biomarkers in anatomic pathology. However, whole-slide imaging (WSI) poses a complex computer vision problem in which the large-scale image resolutions of WSIs and the enormous diversity of morphological phenotypes preclude large-scale data annotation. Current efforts have proposed using pretrained image encoders with either transfer learning from natural image datasets or self-supervised pretraining on publicly-available histopathology datasets, but have not been extensively developed and evaluated across diverse tissue types at scale. We introduce UNI, a general-purpose self-supervised model for pathology, pretrained using over 100 million tissue patches from over 100,000 diagnostic haematoxylin and eosin-stained WSIs across 20 major tissue types, and evaluated on 33 representative CPath clinical tasks in CPath of varying diagnostic difficulties. In addition to outperforming previous state-of-the-art models, we demonstrate new modeling capabilities in CPath such as resolution-agnostic tissue classification, slide classification using few-shot class prototypes, and disease subtyping generalization in classifying up to 108 cancer types in the OncoTree code classification system. UNI advances unsupervised representation learning at scale in CPath in terms of both pretraining data and downstream evaluation, enabling data-efficient AI models that can generalize and transfer to a gamut of diagnostically-challenging tasks and clinical workflows in anatomic pathology.
Modeling Dense Multimodal Interactions Between Biological Pathways and Histology for Survival Prediction
Apr 13, 2023



Integrating whole-slide images (WSIs) and bulk transcriptomics for predicting patient survival can improve our understanding of patient prognosis. However, this multimodal task is particularly challenging due to the different nature of these data: WSIs represent a very high-dimensional spatial description of a tumor, while bulk transcriptomics represent a global description of gene expression levels within that tumor. In this context, our work aims to address two key challenges: (1) how can we tokenize transcriptomics in a semantically meaningful and interpretable way?, and (2) how can we capture dense multimodal interactions between these two modalities? Specifically, we propose to learn biological pathway tokens from transcriptomics that can encode specific cellular functions. Together with histology patch tokens that encode the different morphological patterns in the WSI, we argue that they form appropriate reasoning units for downstream interpretability analyses. We propose fusing both modalities using a memory-efficient multimodal Transformer that can model interactions between pathway and histology patch tokens. Our proposed model, SURVPATH, achieves state-of-the-art performance when evaluated against both unimodal and multimodal baselines on five datasets from The Cancer Genome Atlas. Our interpretability framework identifies key multimodal prognostic factors, and, as such, can provide valuable insights into the interaction between genotype and phenotype, enabling a deeper understanding of the underlying biological mechanisms at play. We make our code public at: https://github.com/ajv012/SurvPath.