 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLLM Agents Making Agent Tools
Feb 17, 2025


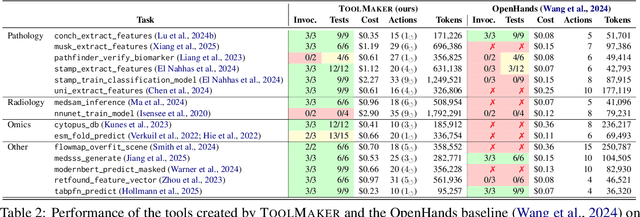
Tool use has turned large language models (LLMs) into powerful agents that can perform complex multi-step tasks by dynamically utilising external software components. However, these tools must be implemented in advance by human developers, hindering the applicability of LLM agents in domains which demand large numbers of highly specialised tools, like in life sciences and medicine. Motivated by the growing trend of scientific studies accompanied by public code repositories, we propose ToolMaker, a novel agentic framework that autonomously transforms papers with code into LLM-compatible tools. Given a short task description and a repository URL, ToolMaker autonomously installs required dependencies and generates code to perform the task, using a closed-loop self-correction mechanism to iteratively diagnose and rectify errors. To evaluate our approach, we introduce a benchmark comprising 15 diverse and complex computational tasks spanning both medical and non-medical domains with over 100 unit tests to objectively assess tool correctness and robustness. ToolMaker correctly implements 80% of the tasks, substantially outperforming current state-of-the-art software engineering agents. ToolMaker therefore is a step towards fully autonomous agent-based scientific workflows.
Abnormality-Driven Representation Learning for Radiology Imaging
Nov 25, 2024



To date, the most common approach for radiology deep learning pipelines is the use of end-to-end 3D networks based on models pre-trained on other tasks, followed by fine-tuning on the task at hand. In contrast, adjacent medical fields such as pathology, which focus on 2D images, have effectively adopted task-agnostic foundational models based on self-supervised learning (SSL), combined with weakly-supervised deep learning (DL). However, the field of radiology still lacks task-agnostic representation models due to the computational and data demands of 3D imaging and the anatomical complexity inherent to radiology scans. To address this gap, we propose CLEAR, a framework for radiology images that uses extracted embeddings from 2D slices along with attention-based aggregation for efficiently predicting clinical endpoints. As part of this framework, we introduce lesion-enhanced contrastive learning (LeCL), a novel approach to obtain visual representations driven by abnormalities in 2D axial slices across different locations of the CT scans. Specifically, we trained single-domain contrastive learning approaches using three different architectures: Vision Transformers, Vision State Space Models and Gated Convolutional Neural Networks. We evaluate our approach across three clinical tasks: tumor lesion location, lung disease detection, and patient staging, benchmarking against four state-of-the-art foundation models, including BiomedCLIP. Our findings demonstrate that CLEAR using representations learned through LeCL, outperforms existing foundation models, while being substantially more compute- and data-efficient.
Unsupervised Foundation Model-Agnostic Slide-Level Representation Learning
Nov 20, 2024



Representation learning of pathology whole-slide images (WSIs) has primarily relied on weak supervision with Multiple Instance Learning (MIL). This approach leads to slide representations highly tailored to a specific clinical task. Self-supervised learning (SSL) has been successfully applied to train histopathology foundation models (FMs) for patch embedding generation. However, generating patient or slide level embeddings remains challenging. Existing approaches for slide representation learning extend the principles of SSL from patch level learning to entire slides by aligning different augmentations of the slide or by utilizing multimodal data. By integrating tile embeddings from multiple FMs, we propose a new single modality SSL method in feature space that generates useful slide representations. Our contrastive pretraining strategy, called COBRA, employs multiple FMs and an architecture based on Mamba-2. COBRA exceeds performance of state-of-the-art slide encoders on four different public CPTAC cohorts on average by at least +3.8% AUC, despite only being pretrained on 3048 WSIs from TCGA. Additionally, COBRA is readily compatible at inference time with previously unseen feature extractors.
End-To-End Clinical Trial Matching with Large Language Models
Jul 18, 2024


Matching cancer patients to clinical trials is essential for advancing treatment and patient care. However, the inconsistent format of medical free text documents and complex trial eligibility criteria make this process extremely challenging and time-consuming for physicians. We investigated whether the entire trial matching process - from identifying relevant trials among 105,600 oncology-related clinical trials on clinicaltrials.gov to generating criterion-level eligibility matches - could be automated using Large Language Models (LLMs). Using GPT-4o and a set of 51 synthetic Electronic Health Records (EHRs), we demonstrate that our approach identifies relevant candidate trials in 93.3% of cases and achieves a preliminary accuracy of 88.0% when matching patient-level information at the criterion level against a baseline defined by human experts. Utilizing LLM feedback reveals that 39.3% criteria that were initially considered incorrect are either ambiguous or inaccurately annotated, leading to a total model accuracy of 92.7% after refining our human baseline. In summary, we present an end-to-end pipeline for clinical trial matching using LLMs, demonstrating high precision in screening and matching trials to individual patients, even outperforming the performance of qualified medical doctors. Our fully end-to-end pipeline can operate autonomously or with human supervision and is not restricted to oncology, offering a scalable solution for enhancing patient-trial matching in real-world settings.
Autonomous Artificial Intelligence Agents for Clinical Decision Making in Oncology
Apr 06, 2024

Multimodal artificial intelligence (AI) systems have the potential to enhance clinical decision-making by interpreting various types of medical data. However, the effectiveness of these models across all medical fields is uncertain. Each discipline presents unique challenges that need to be addressed for optimal performance. This complexity is further increased when attempting to integrate different fields into a single model. Here, we introduce an alternative approach to multimodal medical AI that utilizes the generalist capabilities of a large language model (LLM) as a central reasoning engine. This engine autonomously coordinates and deploys a set of specialized medical AI tools. These tools include text, radiology and histopathology image interpretation, genomic data processing, web searches, and document retrieval from medical guidelines. We validate our system across a series of clinical oncology scenarios that closely resemble typical patient care workflows. We show that the system has a high capability in employing appropriate tools (97%), drawing correct conclusions (93.6%), and providing complete (94%), and helpful (89.2%) recommendations for individual patient cases while consistently referencing relevant literature (82.5%) upon instruction. This work provides evidence that LLMs can effectively plan and execute domain-specific models to retrieve or synthesize new information when used as autonomous agents. This enables them to function as specialist, patient-tailored clinical assistants. It also simplifies regulatory compliance by allowing each component tool to be individually validated and approved. We believe, that our work can serve as a proof-of-concept for more advanced LLM-agents in the medical domain.
In-context learning enables multimodal large language models to classify cancer pathology images
Mar 12, 2024Medical image classification requires labeled, task-specific datasets which are used to train deep learning networks de novo, or to fine-tune foundation models. However, this process is computationally and technically demanding. In language processing, in-context learning provides an alternative, where models learn from within prompts, bypassing the need for parameter updates. Yet, in-context learning remains underexplored in medical image analysis. Here, we systematically evaluate the model Generative Pretrained Transformer 4 with Vision capabilities (GPT-4V) on cancer image processing with in-context learning on three cancer histopathology tasks of high importance: Classification of tissue subtypes in colorectal cancer, colon polyp subtyping and breast tumor detection in lymph node sections. Our results show that in-context learning is sufficient to match or even outperform specialized neural networks trained for particular tasks, while only requiring a minimal number of samples. In summary, this study demonstrates that large vision language models trained on non-domain specific data can be applied out-of-the box to solve medical image-processing tasks in histopathology. This democratizes access of generalist AI models to medical experts without technical background especially for areas where annotated data is scarce.
Joint multi-task learning improves weakly-supervised biomarker prediction in computational pathology
Mar 06, 2024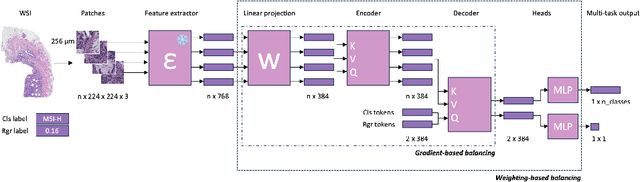


Deep Learning (DL) can predict biomarkers directly from digitized cancer histology in a weakly-supervised setting. Recently, the prediction of continuous biomarkers through regression-based DL has seen an increasing interest. Nonetheless, clinical decision making often requires a categorical outcome. Consequently, we developed a weakly-supervised joint multi-task Transformer architecture which has been trained and evaluated on four public patient cohorts for the prediction of two key predictive biomarkers, microsatellite instability (MSI) and homologous recombination deficiency (HRD), trained with auxiliary regression tasks related to the tumor microenvironment. Moreover, we perform a comprehensive benchmark of 16 approaches of task balancing for weakly-supervised joint multi-task learning in computational pathology. Using our novel approach, we improve over the state-of-the-art area under the receiver operating characteristic by +7.7% and +4.1%, as well as yielding better clustering of latent embeddings by +8% and +5% for the prediction of MSI and HRD in external cohorts, respectively.
From Whole-slide Image to Biomarker Prediction: A Protocol for End-to-End Deep Learning in Computational Pathology
Dec 18, 2023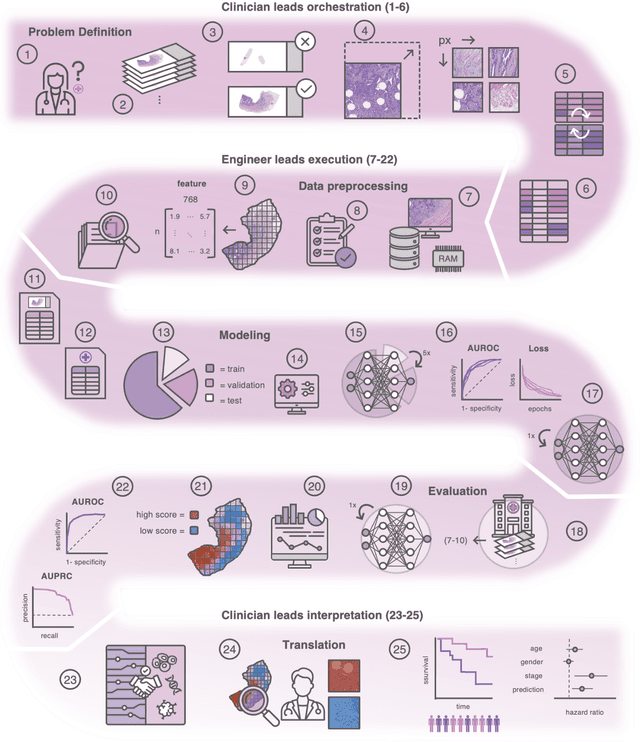
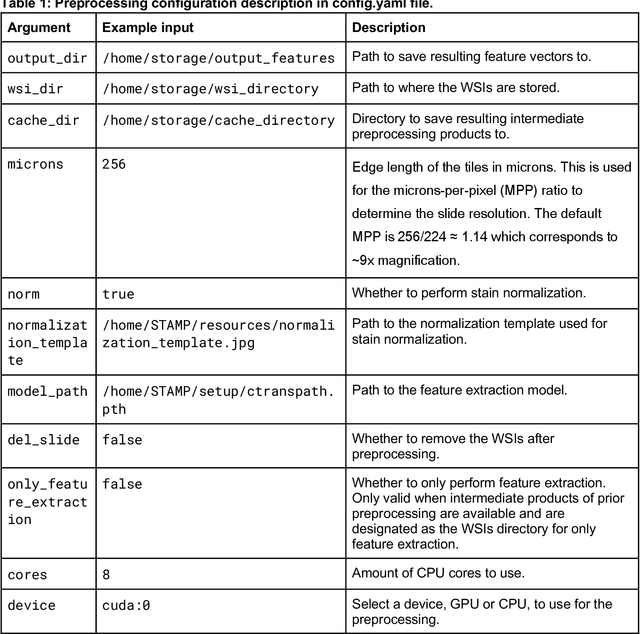
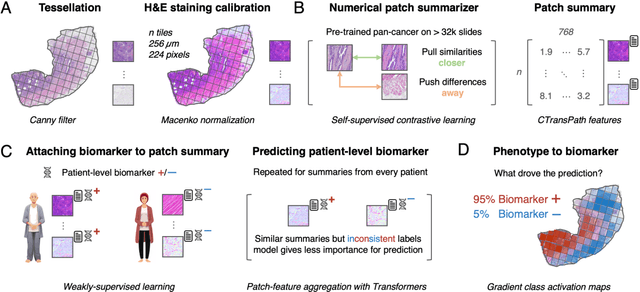

Hematoxylin- and eosin (H&E) stained whole-slide images (WSIs) are the foundation of diagnosis of cancer. In recent years, development of deep learning-based methods in computational pathology enabled the prediction of biomarkers directly from WSIs. However, accurately linking tissue phenotype to biomarkers at scale remains a crucial challenge for democratizing complex biomarkers in precision oncology. This protocol describes a practical workflow for solid tumor associative modeling in pathology (STAMP), enabling prediction of biomarkers directly from WSIs using deep learning. The STAMP workflow is biomarker agnostic and allows for genetic- and clinicopathologic tabular data to be included as an additional input, together with histopathology images. The protocol consists of five main stages which have been successfully applied to various research problems: formal problem definition, data preprocessing, modeling, evaluation and clinical translation. The STAMP workflow differentiates itself through its focus on serving as a collaborative framework that can be used by clinicians and engineers alike for setting up research projects in the field of computational pathology. As an example task, we applied STAMP to the prediction of microsatellite instability (MSI) status in colorectal cancer, showing accurate performance for the identification of MSI-high tumors. Moreover, we provide an open-source codebase which has been deployed at several hospitals across the globe to set up computational pathology workflows. The STAMP workflow requires one workday of hands-on computational execution and basic command line knowledge.
A Good Feature Extractor Is All You Need for Weakly Supervised Learning in Histopathology
Nov 29, 2023


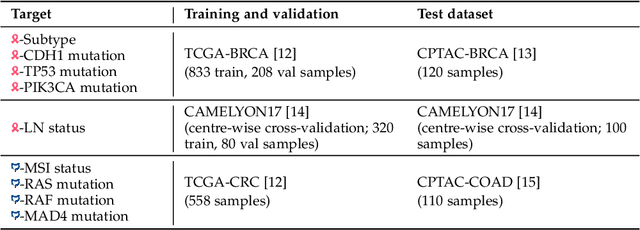
Deep learning is revolutionising pathology, offering novel opportunities in disease prognosis and personalised treatment. Historically, stain normalisation has been a crucial preprocessing step in computational pathology pipelines, and persists into the deep learning era. Yet, with the emergence of feature extractors trained using self-supervised learning (SSL) on diverse pathology datasets, we call this practice into question. In an empirical evaluation of publicly available feature extractors, we find that omitting stain normalisation and image augmentations does not compromise downstream performance, while incurring substantial savings in memory and compute. Further, we show that the top-performing feature extractors are remarkably robust to variations in stain and augmentations like rotation in their latent space. Contrary to previous patch-level benchmarking studies, our approach emphasises clinical relevance by focusing on slide-level prediction tasks in a weakly supervised setting with external validation cohorts. This work represents the most comprehensive robustness evaluation of public pathology SSL feature extractors to date, involving more than 6,000 training runs across nine tasks, five datasets, three downstream architectures, and various preprocessing setups. Our findings stand to streamline digital pathology workflows by minimising preprocessing needs and informing the selection of feature extractors.
Deep Multiple Instance Learning with Distance-Aware Self-Attention
May 20, 2023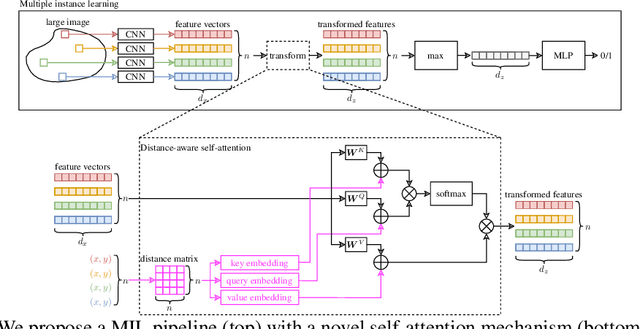
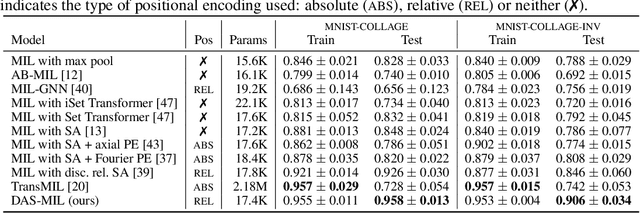

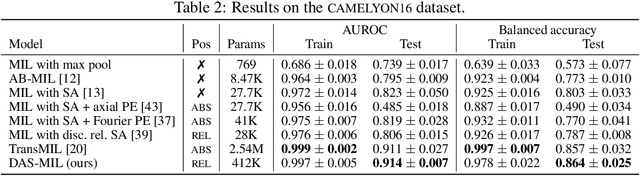
Traditional supervised learning tasks require a label for every instance in the training set, but in many real-world applications, labels are only available for collections (bags) of instances. This problem setting, known as multiple instance learning (MIL), is particularly relevant in the medical domain, where high-resolution images are split into smaller patches, but labels apply to the image as a whole. Recent MIL models are able to capture correspondences between patches by employing self-attention, allowing them to weigh each patch differently based on all other patches in the bag. However, these approaches still do not consider the relative spatial relationships between patches within the larger image, which is especially important in computational pathology. To this end, we introduce a novel MIL model with distance-aware self-attention (DAS-MIL), which explicitly takes into account relative spatial information when modelling the interactions between patches. Unlike existing relative position representations for self-attention which are discrete, our approach introduces continuous distance-dependent terms into the computation of the attention weights, and is the first to apply relative position representations in the context of MIL. We evaluate our model on a custom MNIST-based MIL dataset that requires the consideration of relative spatial information, as well as on CAMELYON16, a publicly available cancer metastasis detection dataset, where we achieve a test AUROC score of 0.91. On both datasets, our model outperforms existing MIL approaches that employ absolute positional encodings, as well as existing relative position representation schemes applied to MIL. Our code is available at https://anonymous.4open.science/r/das-mil.