 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFrom Whole-slide Image to Biomarker Prediction: A Protocol for End-to-End Deep Learning in Computational Pathology
Dec 18, 2023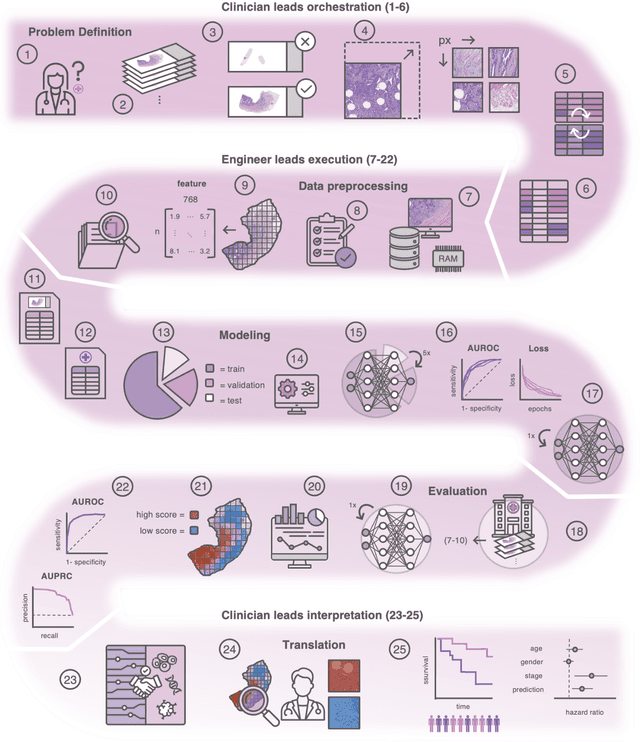
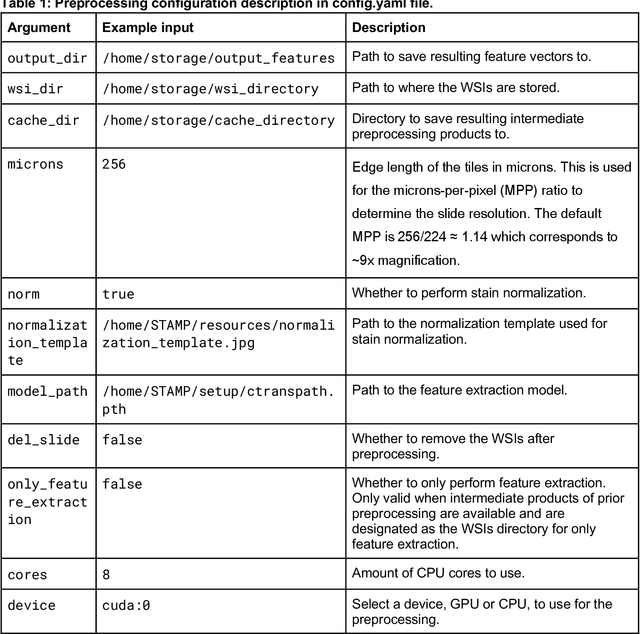
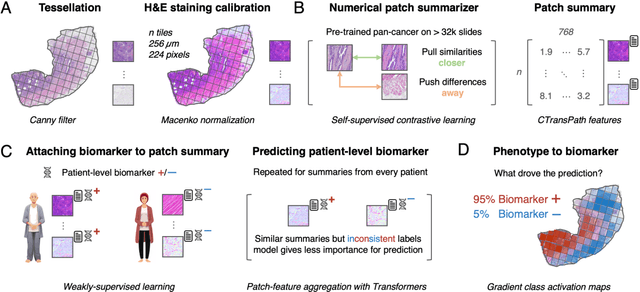

Hematoxylin- and eosin (H&E) stained whole-slide images (WSIs) are the foundation of diagnosis of cancer. In recent years, development of deep learning-based methods in computational pathology enabled the prediction of biomarkers directly from WSIs. However, accurately linking tissue phenotype to biomarkers at scale remains a crucial challenge for democratizing complex biomarkers in precision oncology. This protocol describes a practical workflow for solid tumor associative modeling in pathology (STAMP), enabling prediction of biomarkers directly from WSIs using deep learning. The STAMP workflow is biomarker agnostic and allows for genetic- and clinicopathologic tabular data to be included as an additional input, together with histopathology images. The protocol consists of five main stages which have been successfully applied to various research problems: formal problem definition, data preprocessing, modeling, evaluation and clinical translation. The STAMP workflow differentiates itself through its focus on serving as a collaborative framework that can be used by clinicians and engineers alike for setting up research projects in the field of computational pathology. As an example task, we applied STAMP to the prediction of microsatellite instability (MSI) status in colorectal cancer, showing accurate performance for the identification of MSI-high tumors. Moreover, we provide an open-source codebase which has been deployed at several hospitals across the globe to set up computational pathology workflows. The STAMP workflow requires one workday of hands-on computational execution and basic command line knowledge.
Regression-based Deep-Learning predicts molecular biomarkers from pathology slides
Apr 11, 2023Deep Learning (DL) can predict biomarkers from cancer histopathology. Several clinically approved applications use this technology. Most approaches, however, predict categorical labels, whereas biomarkers are often continuous measurements. We hypothesized that regression-based DL outperforms classification-based DL. Therefore, we developed and evaluated a new self-supervised attention-based weakly supervised regression method that predicts continuous biomarkers directly from images in 11,671 patients across nine cancer types. We tested our method for multiple clinically and biologically relevant biomarkers: homologous repair deficiency (HRD) score, a clinically used pan-cancer biomarker, as well as markers of key biological processes in the tumor microenvironment. Using regression significantly enhances the accuracy of biomarker prediction, while also improving the interpretability of the results over classification. In a large cohort of colorectal cancer patients, regression-based prediction scores provide a higher prognostic value than classification-based scores. Our open-source regression approach offers a promising alternative for continuous biomarker analysis in computational pathology.
Test Time Transform Prediction for Open Set Histopathological Image Recognition
Jun 27, 2022
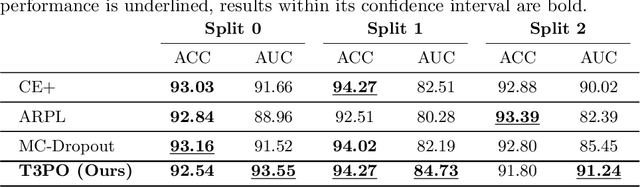
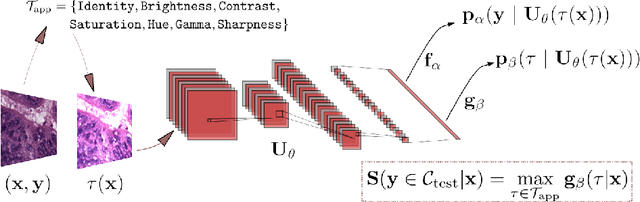
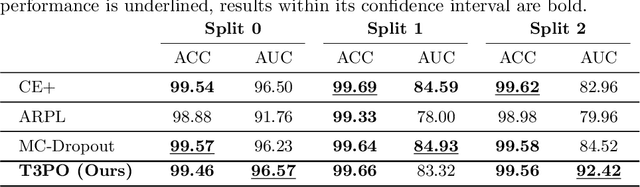
Tissue typology annotation in Whole Slide histological images is a complex and tedious, yet necessary task for the development of computational pathology models. We propose to address this problem by applying Open Set Recognition techniques to the task of jointly classifying tissue that belongs to a set of annotated classes, e.g. clinically relevant tissue categories, while rejecting in test time Open Set samples, i.e. images that belong to categories not present in the training set. To this end, we introduce a new approach for Open Set histopathological image recognition based on training a model to accurately identify image categories and simultaneously predict which data augmentation transform has been applied. In test time, we measure model confidence in predicting this transform, which we expect to be lower for images in the Open Set. We carry out comprehensive experiments in the context of colorectal cancer assessment from histological images, which provide evidence on the strengths of our approach to automatically identify samples from unknown categories. Code is released at https://github.com/agaldran/t3po .