 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Standardized Framework For Evaluating Gene Expression Generative Models
Mar 11, 2026The rapid development of generative models for single-cell gene expression data has created an urgent need for standardised evaluation frameworks. Current evaluation practices suffer from inconsistent metric implementations, incomparable hyperparameter choices, and a lack of biologically-grounded metrics. We present Generated Genetic Expression Evaluator (GGE), an open-source Python framework that addresses these challenges by providing a comprehensive suite of distributional metrics with explicit computation space options and biologically-motivated evaluation through differentially expressed gene (DEG)-focused analysis and perturbation-effect correlation, enabling standardized reporting and reproducible benchmarking. Through extensive analysis of the single-cell generative modeling literature, we identify that no standardized evaluation protocol exists. Methods report incomparable metrics computed in different spaces with different hyperparameters. We demonstrate that metric values vary substantially depending on implementation choices, highlighting the critical need for standardization. GGE enables fair comparison across generative approaches and accelerates progress in perturbation response prediction, cellular identity modeling, and counterfactual inference.
DirMoE: Dirichlet-routed Mixture of Experts
Feb 09, 2026Mixture-of-Experts (MoE) models have demonstrated exceptional performance in large-scale language models. Existing routers typically rely on non-differentiable Top-$k$+Softmax, limiting their performance and scalability. We argue that two distinct decisions, which experts to activate and how to distribute expert contributions among them, are conflated in standard Top-$k$+Softmax. We introduce Dirichlet-Routed MoE (DirMoE), a novel end-to-end differentiable routing mechanism built on a Dirichlet variational autoencoder framework. This design fundamentally disentangles the core routing problems: expert selection, modeled by a Bernoulli component, and expert contribution among chosen experts, handled by a Dirichlet component. The entire forward pass remains fully differentiable through the use of Gumbel-Sigmoid relaxation for the expert selection and implicit reparameterization for the Dirichlet distribution. Our training objective, a variational ELBO, includes a direct sparsity penalty that precisely controls the number of active experts in expectation, alongside a schedule for key hyperparameters that guides the model from an exploratory to a definitive routing state. Moreover, our DirMoE router matches or exceeds other methods while improving expert specialization.
SIGMMA: Hierarchical Graph-Based Multi-Scale Multi-modal Contrastive Alignment of Histopathology Image and Spatial Transcriptome
Nov 19, 2025



Recent advances in computational pathology have leveraged vision-language models to learn joint representations of Hematoxylin and Eosin (HE) images with spatial transcriptomic (ST) profiles. However, existing approaches typically align HE tiles with their corresponding ST profiles at a single scale, overlooking fine-grained cellular structures and their spatial organization. To address this, we propose Sigmma, a multi-modal contrastive alignment framework for learning hierarchical representations of HE images and spatial transcriptome profiles across multiple scales. Sigmma introduces multi-scale contrastive alignment, ensuring that representations learned at different scales remain coherent across modalities. Furthermore, by representing cell interactions as a graph and integrating inter- and intra-subgraph relationships, our approach effectively captures cell-cell interactions, ranging from fine to coarse, within the tissue microenvironment. We demonstrate that Sigmm learns representations that better capture cross-modal correspondences, leading to an improvement of avg. 9.78\% in the gene-expression prediction task and avg. 26.93\% in the cross-modal retrieval task across datasets. We further show that it learns meaningful multi-tissue organization in downstream analyses.
3D-Guided Scalable Flow Matching for Generating Volumetric Tissue Spatial Transcriptomics from Serial Histology
Nov 18, 2025A scalable and robust 3D tissue transcriptomics profile can enable a holistic understanding of tissue organization and provide deeper insights into human biology and disease. Most predictive algorithms that infer ST directly from histology treat each section independently and ignore 3D structure, while existing 3D-aware approaches are not generative and do not scale well. We present Holographic Tissue Expression Inpainting and Analysis (HoloTea), a 3D-aware flow-matching framework that imputes spot-level gene expression from H&E while explicitly using information from adjacent sections. Our key idea is to retrieve morphologically corresponding spots on neighboring slides in a shared feature space and fuse this cross section context into a lightweight ControlNet, allowing conditioning to follow anatomical continuity. To better capture the count nature of the data, we introduce a 3D-consistent prior for flow matching that combines a learned zero-inflated negative binomial (ZINB) prior with a spatial-empirical prior constructed from neighboring sections. A global attention block introduces 3D H&E scaling linearly with the number of spots in the slide, enabling training and inference on large 3D ST datasets. Across three spatial transcriptomics datasets spanning different tissue types and resolutions, HoloTea consistently improves 3D expression accuracy and generalization compared to 2D and 3D baselines. We envision HoloTea advancing the creation of accurate 3D virtual tissues, ultimately accelerating biomarker discovery and deepening our understanding of disease.
Predicting single-cell perturbation responses for unseen drugs
Apr 28, 2022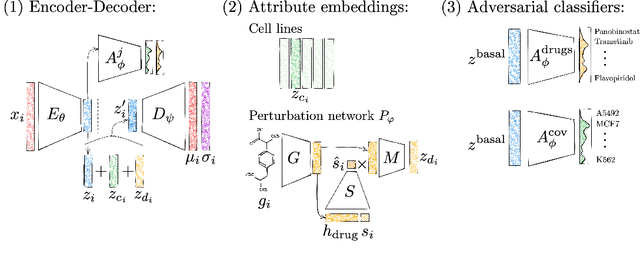
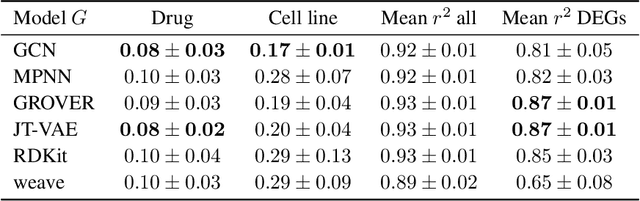
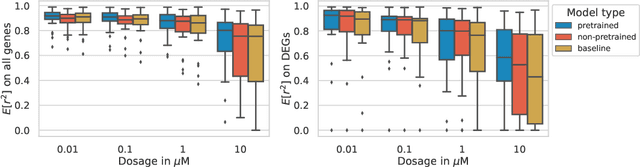
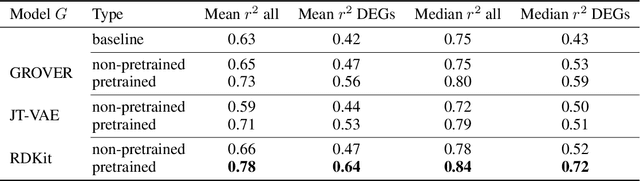
Single-cell transcriptomics enabled the study of cellular heterogeneity in response to perturbations at the resolution of individual cells. However, scaling high-throughput screens (HTSs) to measure cellular responses for many drugs remains a challenge due to technical limitations and, more importantly, the cost of such multiplexed experiments. Thus, transferring information from routinely performed bulk RNA-seq HTS is required to enrich single-cell data meaningfully. We introduce a new encoder-decoder architecture to study the perturbational effects of unseen drugs. We combine the model with a transfer learning scheme and demonstrate how training on existing bulk RNA-seq HTS datasets can improve generalisation performance. Better generalisation reduces the need for extensive and costly screens at single-cell resolution. We envision that our proposed method will facilitate more efficient experiment designs through its ability to generate in-silico hypotheses, ultimately accelerating targeted drug discovery.
* 8 pages
Conditional out-of-sample generation for unpaired data using trVAE
Oct 30, 2019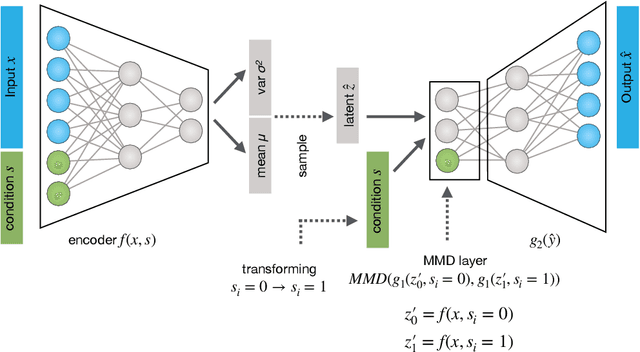
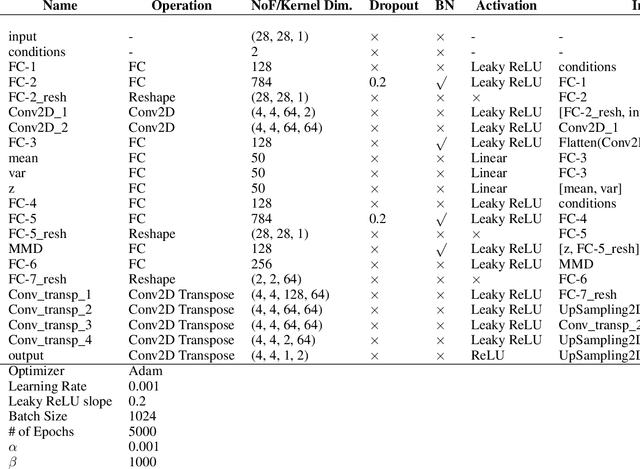
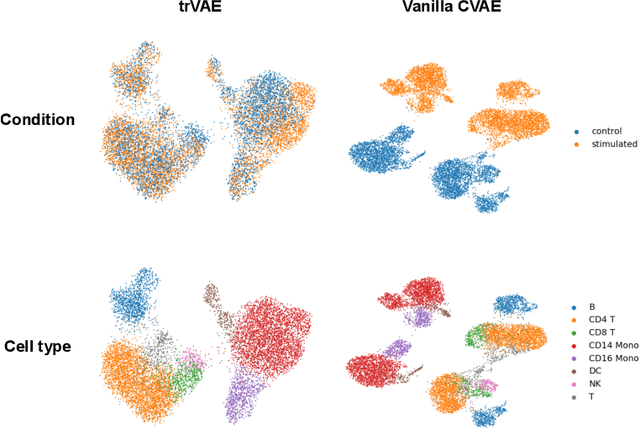
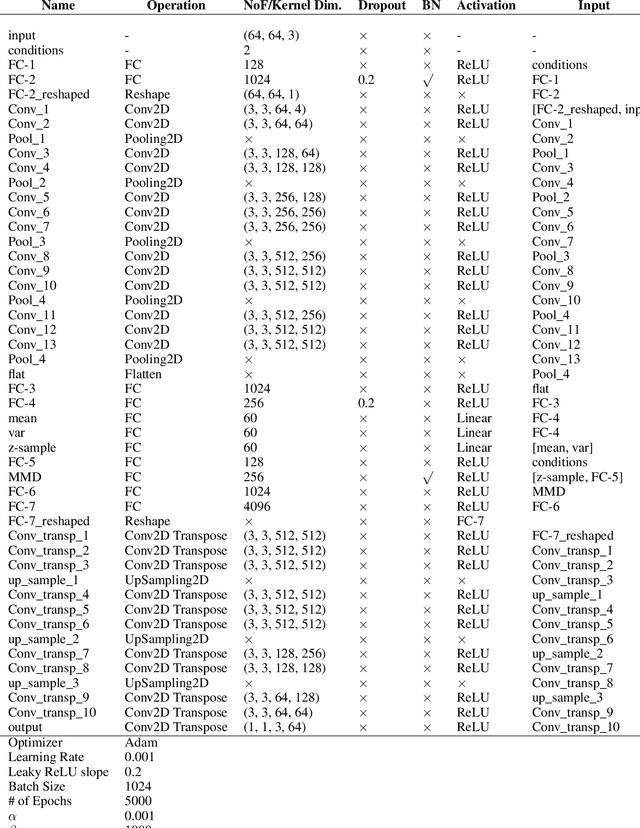
While generative models have shown great success in generating high-dimensional samples conditional on low-dimensional descriptors (learning e.g. stroke thickness in MNIST, hair color in CelebA, or speaker identity in Wavenet), their generation out-of-sample poses fundamental problems. The conditional variational autoencoder (CVAE) as a simple conditional generative model does not explicitly relate conditions during training and, hence, has no incentive of learning a compact joint distribution across conditions. We overcome this limitation by matching their distributions using maximum mean discrepancy (MMD) in the decoder layer that follows the bottleneck. This introduces a strong regularization both for reconstructing samples within the same condition and for transforming samples across conditions, resulting in much improved generalization. We refer to the architecture as \emph{transformer} VAE (trVAE). Benchmarking trVAE on high-dimensional image and tabular data, we demonstrate higher robustness and higher accuracy than existing approaches. In particular, we show qualitatively improved predictions for cellular perturbation response to treatment and disease based on high-dimensional single-cell gene expression data, by tackling previously problematic minority classes and multiple conditions. For generic tasks, we improve Pearson correlations of high-dimensional estimated means and variances with their ground truths from 0.89 to 0.97 and 0.75 to 0.87, respectively.
Deep Packet: A Novel Approach For Encrypted Traffic Classification Using Deep Learning
Jul 04, 2018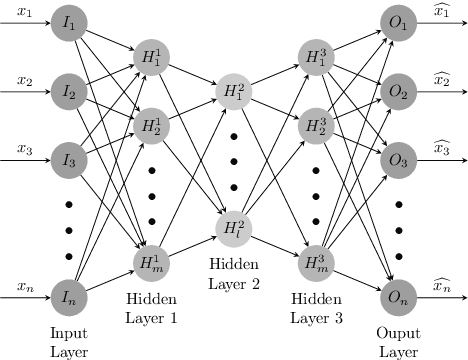
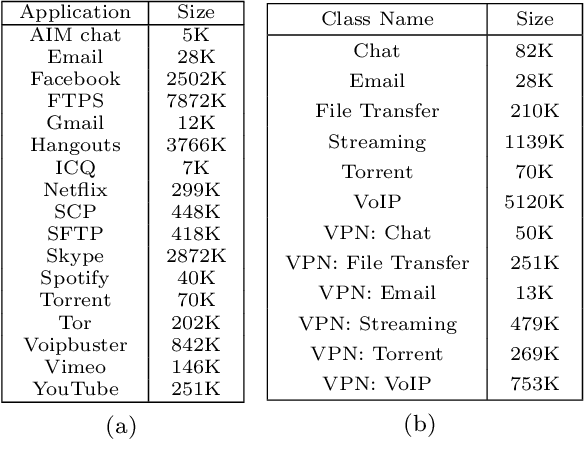
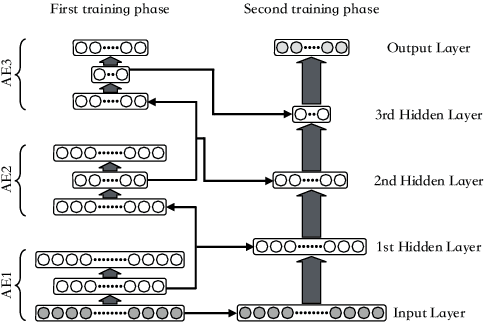
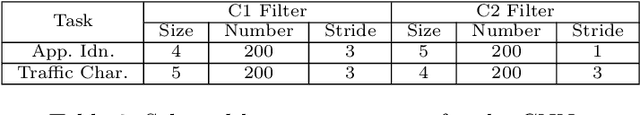
Internet traffic classification has become more important with rapid growth of current Internet network and online applications. There have been numerous studies on this topic which have led to many different approaches. Most of these approaches use predefined features extracted by an expert in order to classify network traffic. In contrast, in this study, we propose a \emph{deep learning} based approach which integrates both feature extraction and classification phases into one system. Our proposed scheme, called "Deep Packet," can handle both \emph{traffic characterization} in which the network traffic is categorized into major classes (\eg, FTP and P2P) and application identification in which end-user applications (\eg, BitTorrent and Skype) identification is desired. Contrary to most of the current methods, Deep Packet can identify encrypted traffic and also distinguishes between VPN and non-VPN network traffic. After an initial pre-processing phase on data, packets are fed into Deep Packet framework that embeds stacked autoencoder and convolution neural network in order to classify network traffic. Deep packet with CNN as its classification model achieved recall of $0.98$ in application identification task and $0.94$ in traffic categorization task. To the best of our knowledge, Deep Packet outperforms all of the proposed classification methods on UNB ISCX VPN-nonVPN dataset.