 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeConditionally Invariant Representation Learning for Disentangling Cellular Heterogeneity
Paper and Code
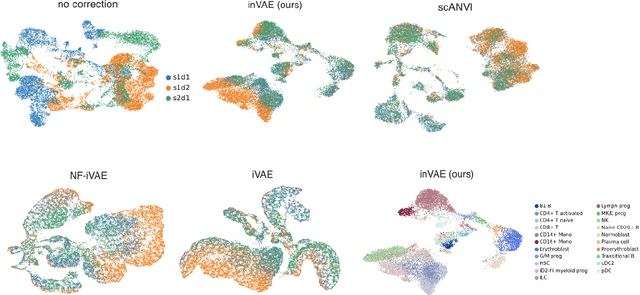
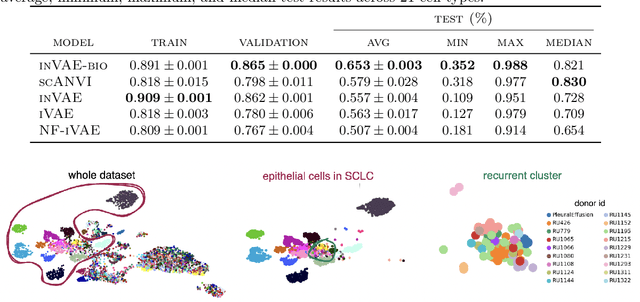
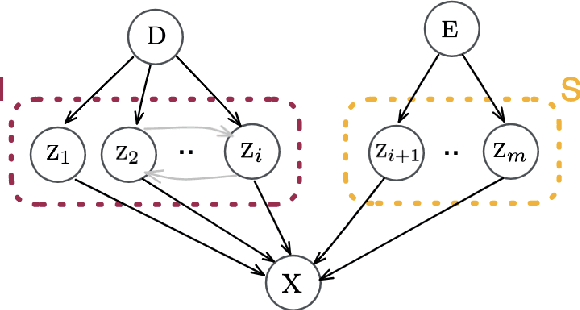
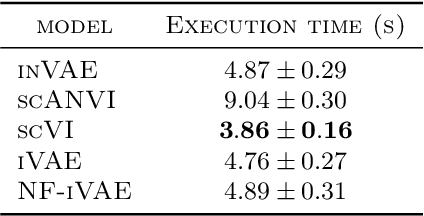
This paper presents a novel approach that leverages domain variability to learn representations that are conditionally invariant to unwanted variability or distractors. Our approach identifies both spurious and invariant latent features necessary for achieving accurate reconstruction by placing distinct conditional priors on latent features. The invariant signals are disentangled from noise by enforcing independence which facilitates the construction of an interpretable model with a causal semantic. By exploiting the interplay between data domains and labels, our method simultaneously identifies invariant features and builds invariant predictors. We apply our method to grand biological challenges, such as data integration in single-cell genomics with the aim of capturing biological variations across datasets with many samples, obtained from different conditions or multiple laboratories. Our approach allows for the incorporation of specific biological mechanisms, including gene programs, disease states, or treatment conditions into the data integration process, bridging the gap between the theoretical assumptions and real biological applications. Specifically, the proposed approach helps to disentangle biological signals from data biases that are unrelated to the target task or the causal explanation of interest. Through extensive benchmarking using large-scale human hematopoiesis and human lung cancer data, we validate the superiority of our approach over existing methods and demonstrate that it can empower deeper insights into cellular heterogeneity and the identification of disease cell states.