 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to Edgecubic: CUDA-accelerated 3D Bioimage Computing
Oct 15, 2025Quantitative analysis of multidimensional biological images is useful for understanding complex cellular phenotypes and accelerating advances in biomedical research. As modern microscopy generates ever-larger 2D and 3D datasets, existing computational approaches are increasingly limited by their scalability, efficiency, and integration with modern scientific computing workflows. Existing bioimage analysis tools often lack application programmable interfaces (APIs), do not support graphics processing unit (GPU) acceleration, lack broad 3D image processing capabilities, and/or have poor interoperability for compute-heavy workflows. Here, we introduce cubic, an open-source Python library that addresses these challenges by augmenting widely used SciPy and scikit-image APIs with GPU-accelerated alternatives from CuPy and RAPIDS cuCIM. cubic's API is device-agnostic and dispatches operations to GPU when data reside on the device and otherwise executes on CPU, seamlessly accelerating a broad range of image processing routines. This approach enables GPU acceleration of existing bioimage analysis workflows, from preprocessing to segmentation and feature extraction for 2D and 3D data. We evaluate cubic both by benchmarking individual operations and by reproducing existing deconvolution and segmentation pipelines, achieving substantial speedups while maintaining algorithmic fidelity. These advances establish a robust foundation for scalable, reproducible bioimage analysis that integrates with the broader Python scientific computing ecosystem, including other GPU-accelerated methods, enabling both interactive exploration and automated high-throughput analysis workflows. cubic is openly available at https://github$.$com/alxndrkalinin/cubic
cp_measure: API-first feature extraction for image-based profiling workflows
Jul 01, 2025Biological image analysis has traditionally focused on measuring specific visual properties of interest for cells or other entities. A complementary paradigm gaining increasing traction is image-based profiling - quantifying many distinct visual features to form comprehensive profiles which may reveal hidden patterns in cellular states, drug responses, and disease mechanisms. While current tools like CellProfiler can generate these feature sets, they pose significant barriers to automated and reproducible analyses, hindering machine learning workflows. Here we introduce cp_measure, a Python library that extracts CellProfiler's core measurement capabilities into a modular, API-first tool designed for programmatic feature extraction. We demonstrate that cp_measure features retain high fidelity with CellProfiler features while enabling seamless integration with the scientific Python ecosystem. Through applications to 3D astrocyte imaging and spatial transcriptomics, we showcase how cp_measure enables reproducible, automated image-based profiling pipelines that scale effectively for machine learning applications in computational biology.
Learning Molecular Representation in a Cell
Jun 17, 2024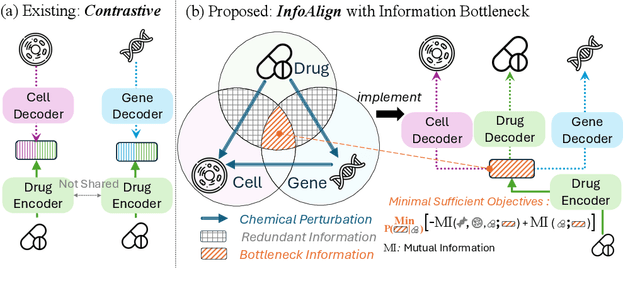
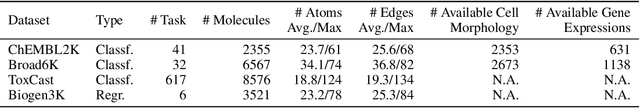
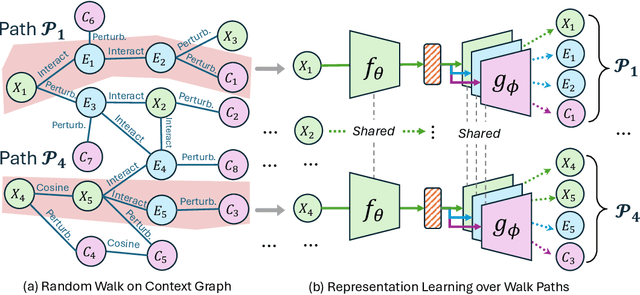
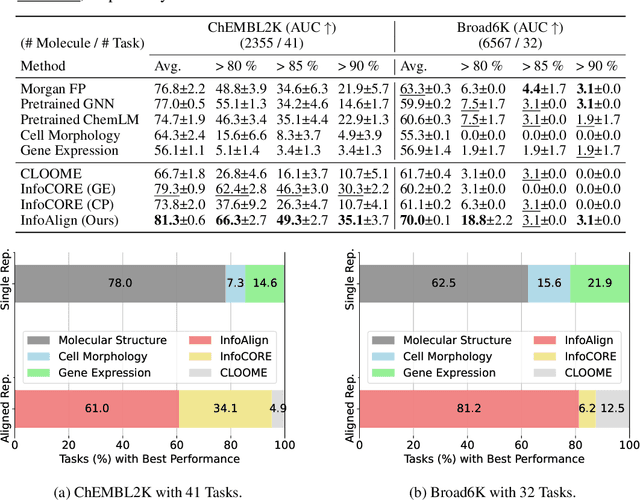
Predicting drug efficacy and safety in vivo requires information on biological responses (e.g., cell morphology and gene expression) to small molecule perturbations. However, current molecular representation learning methods do not provide a comprehensive view of cell states under these perturbations and struggle to remove noise, hindering model generalization. We introduce the Information Alignment (InfoAlign) approach to learn molecular representations through the information bottleneck method in cells. We integrate molecules and cellular response data as nodes into a context graph, connecting them with weighted edges based on chemical, biological, and computational criteria. For each molecule in a training batch, InfoAlign optimizes the encoder's latent representation with a minimality objective to discard redundant structural information. A sufficiency objective decodes the representation to align with different feature spaces from the molecule's neighborhood in the context graph. We demonstrate that the proposed sufficiency objective for alignment is tighter than existing encoder-based contrastive methods. Empirically, we validate representations from InfoAlign in two downstream tasks: molecular property prediction against up to 19 baseline methods across four datasets, plus zero-shot molecule-morphology matching.
Pseudo-Labeling Enhanced by Privileged Information and Its Application to In Situ Sequencing Images
Jun 28, 2023



Various strategies for label-scarce object detection have been explored by the computer vision research community. These strategies mainly rely on assumptions that are specific to natural images and not directly applicable to the biological and biomedical vision domains. For example, most semi-supervised learning strategies rely on a small set of labeled data as a confident source of ground truth. In many biological vision applications, however, the ground truth is unknown and indirect information might be available in the form of noisy estimations or orthogonal evidence. In this work, we frame a crucial problem in spatial transcriptomics - decoding barcodes from In-Situ-Sequencing (ISS) images - as a semi-supervised object detection (SSOD) problem. Our proposed framework incorporates additional available sources of information into a semi-supervised learning framework in the form of privileged information. The privileged information is incorporated into the teacher's pseudo-labeling in a teacher-student self-training iteration. Although the available privileged information could be data domain specific, we have introduced a general strategy of pseudo-labeling enhanced by privileged information (PLePI) and exemplified the concept using ISS images, as well on the COCO benchmark using extra evidence provided by CLIP.
* This paper has been accepted for publication at IJCAI 2023
Towards automated high-throughput screening of C. elegans on agar
Mar 22, 2010



High-throughput screening (HTS) using model organisms is a promising method to identify a small number of genes or drugs potentially relevant to human biology or disease. In HTS experiments, robots and computers do a significant portion of the experimental work. However, one remaining major bottleneck is the manual analysis of experimental results, which is commonly in the form of microscopy images. This manual inspection is labor intensive, slow and subjective. Here we report our progress towards applying computer vision and machine learning methods to analyze HTS experiments that use Caenorhabditis elegans (C. elegans) worms grown on agar. Our main contribution is a robust segmentation algorithm for separating the worms from the background using brightfield images. We also show that by combining the output of this segmentation algorithm with an algorithm to detect the fluorescent dye, Nile Red, we can reliably distinguish different fluorescence-based phenotypes even though the visual differences are subtle. The accuracy of our method is similar to that of expert human analysts. This new capability is a significant step towards fully automated HTS experiments using C. elegans.