 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAI Agents in Drug Discovery
Oct 31, 2025Artificial intelligence (AI) agents are emerging as transformative tools in drug discovery, with the ability to autonomously reason, act, and learn through complicated research workflows. Building on large language models (LLMs) coupled with perception, computation, action, and memory tools, these agentic AI systems could integrate diverse biomedical data, execute tasks, carry out experiments via robotic platforms, and iteratively refine hypotheses in closed loops. We provide a conceptual and technical overview of agentic AI architectures, ranging from ReAct and Reflection to Supervisor and Swarm systems, and illustrate their applications across key stages of drug discovery, including literature synthesis, toxicity prediction, automated protocol generation, small-molecule synthesis, drug repurposing, and end-to-end decision-making. To our knowledge, this represents the first comprehensive work to present real-world implementations and quantifiable impacts of agentic AI systems deployed in operational drug discovery settings. Early implementations demonstrate substantial gains in speed, reproducibility, and scalability, compressing workflows that once took months into hours while maintaining scientific traceability. We discuss the current challenges related to data heterogeneity, system reliability, privacy, and benchmarking, and outline future directions towards technology in support of science and translation.
Learning Molecular Representation in a Cell
Jun 17, 2024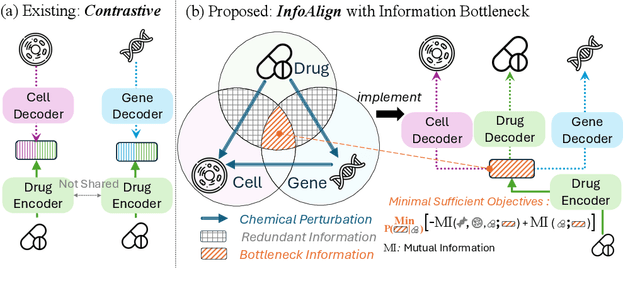
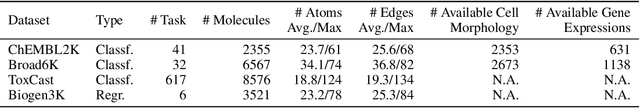
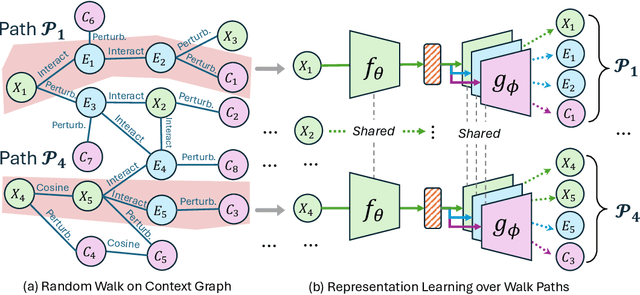
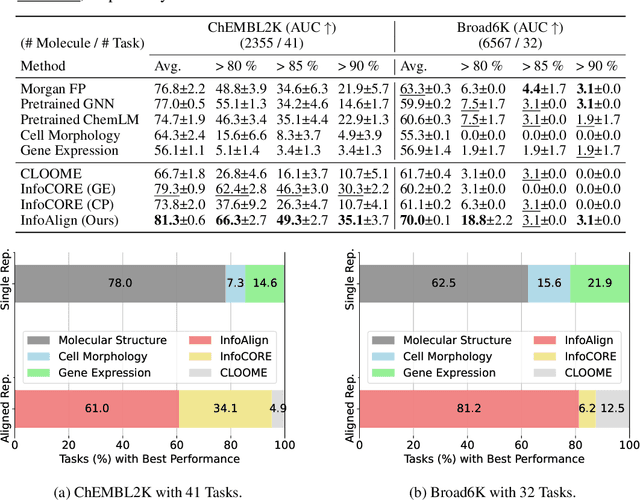
Predicting drug efficacy and safety in vivo requires information on biological responses (e.g., cell morphology and gene expression) to small molecule perturbations. However, current molecular representation learning methods do not provide a comprehensive view of cell states under these perturbations and struggle to remove noise, hindering model generalization. We introduce the Information Alignment (InfoAlign) approach to learn molecular representations through the information bottleneck method in cells. We integrate molecules and cellular response data as nodes into a context graph, connecting them with weighted edges based on chemical, biological, and computational criteria. For each molecule in a training batch, InfoAlign optimizes the encoder's latent representation with a minimality objective to discard redundant structural information. A sufficiency objective decodes the representation to align with different feature spaces from the molecule's neighborhood in the context graph. We demonstrate that the proposed sufficiency objective for alignment is tighter than existing encoder-based contrastive methods. Empirically, we validate representations from InfoAlign in two downstream tasks: molecular property prediction against up to 19 baseline methods across four datasets, plus zero-shot molecule-morphology matching.
Understanding Biology in the Age of Artificial Intelligence
Mar 06, 2024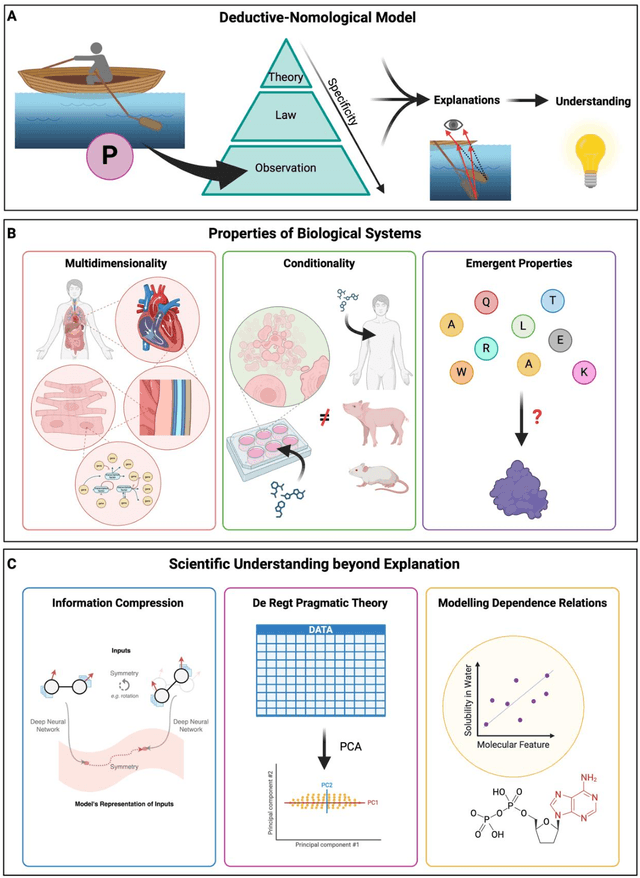
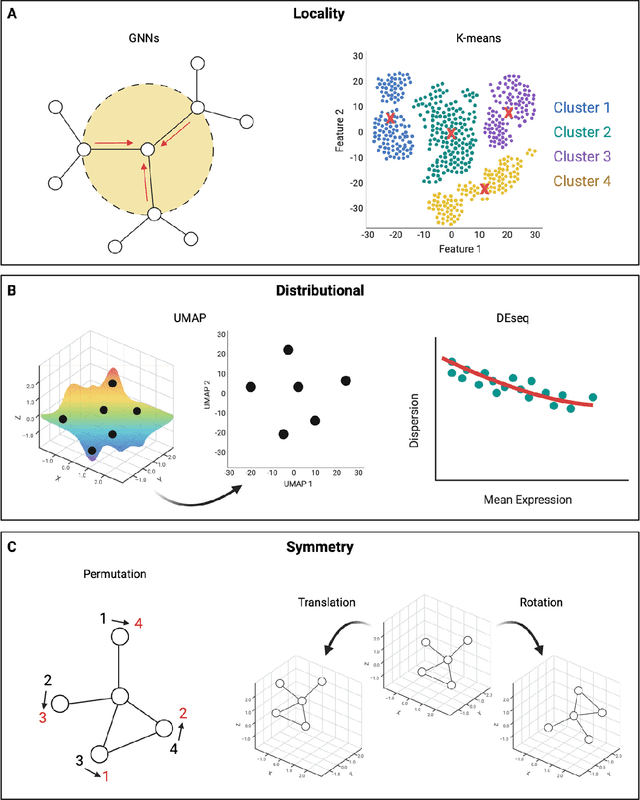
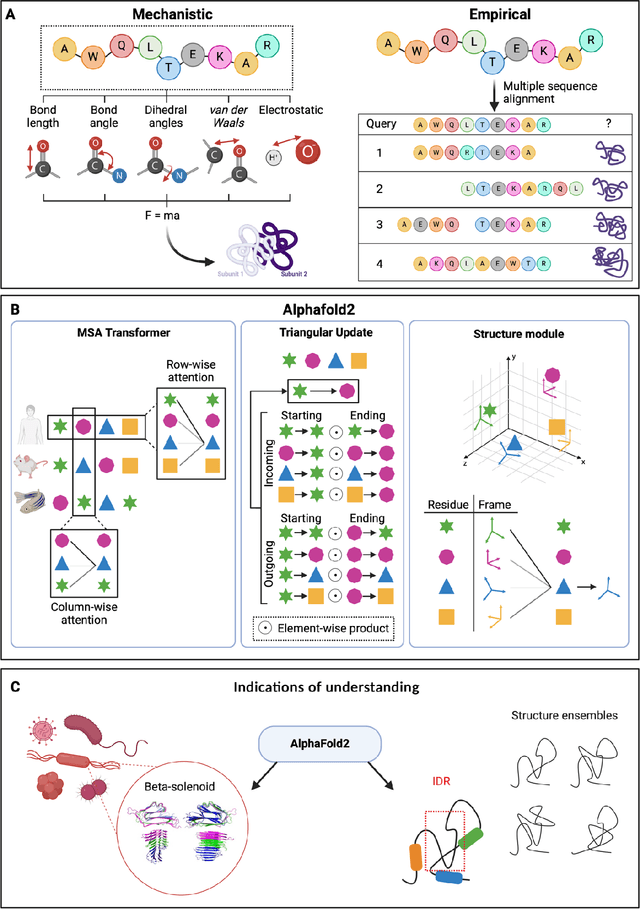
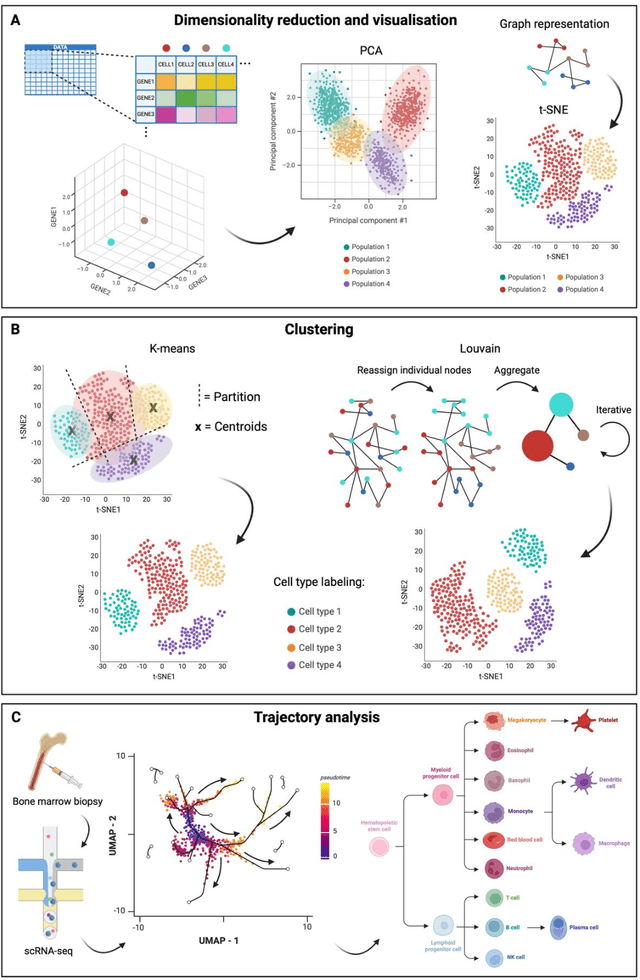
Modern life sciences research is increasingly relying on artificial intelligence approaches to model biological systems, primarily centered around the use of machine learning (ML) models. Although ML is undeniably useful for identifying patterns in large, complex data sets, its widespread application in biological sciences represents a significant deviation from traditional methods of scientific inquiry. As such, the interplay between these models and scientific understanding in biology is a topic with important implications for the future of scientific research, yet it is a subject that has received little attention. Here, we draw from an epistemological toolkit to contextualize recent applications of ML in biological sciences under modern philosophical theories of understanding, identifying general principles that can guide the design and application of ML systems to model biological phenomena and advance scientific knowledge. We propose that conceptions of scientific understanding as information compression, qualitative intelligibility, and dependency relation modelling provide a useful framework for interpreting ML-mediated understanding of biological systems. Through a detailed analysis of two key application areas of ML in modern biological research - protein structure prediction and single cell RNA-sequencing - we explore how these features have thus far enabled ML systems to advance scientific understanding of their target phenomena, how they may guide the development of future ML models, and the key obstacles that remain in preventing ML from achieving its potential as a tool for biological discovery. Consideration of the epistemological features of ML applications in biology will improve the prospects of these methods to solve important problems and advance scientific understanding of living systems.