 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeIgCraft: A versatile sequence generation framework for antibody discovery and engineering
Mar 25, 2025



Designing antibody sequences to better resemble those observed in natural human repertoires is a key challenge in biologics development. We introduce IgCraft: a multi-purpose model for paired human antibody sequence generation, built on Bayesian Flow Networks. IgCraft presents one of the first unified generative modeling frameworks capable of addressing multiple antibody sequence design tasks with a single model, including unconditional sampling, sequence inpainting, inverse folding, and CDR motif scaffolding. Our approach achieves competitive results across the full spectrum of these tasks while constraining generation to the space of human antibody sequences, exhibiting particular strengths in CDR motif scaffolding (grafting) where we achieve state-of-the-art performance in terms of humanness and preservation of structural properties. By integrating previously separate tasks into a single scalable generative model, IgCraft provides a versatile platform for sampling human antibody sequences under a variety of contexts relevant to antibody discovery and engineering. Model code and weights are publicly available at github.com/mgreenig/IgCraft.
Understanding Biology in the Age of Artificial Intelligence
Mar 06, 2024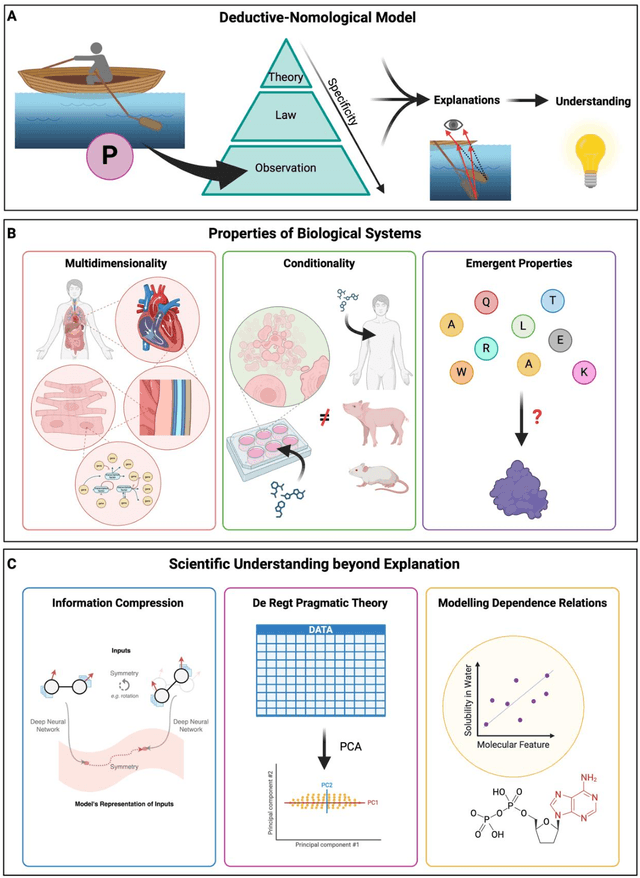
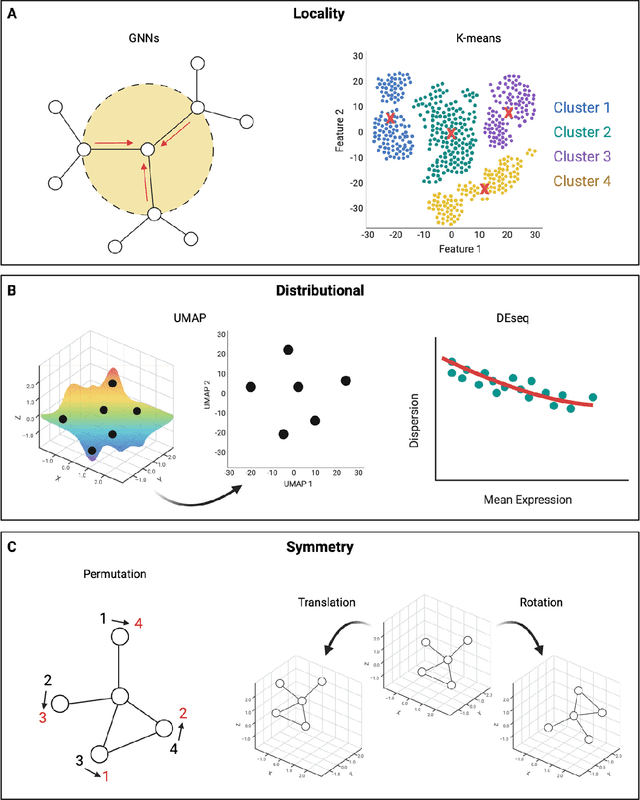
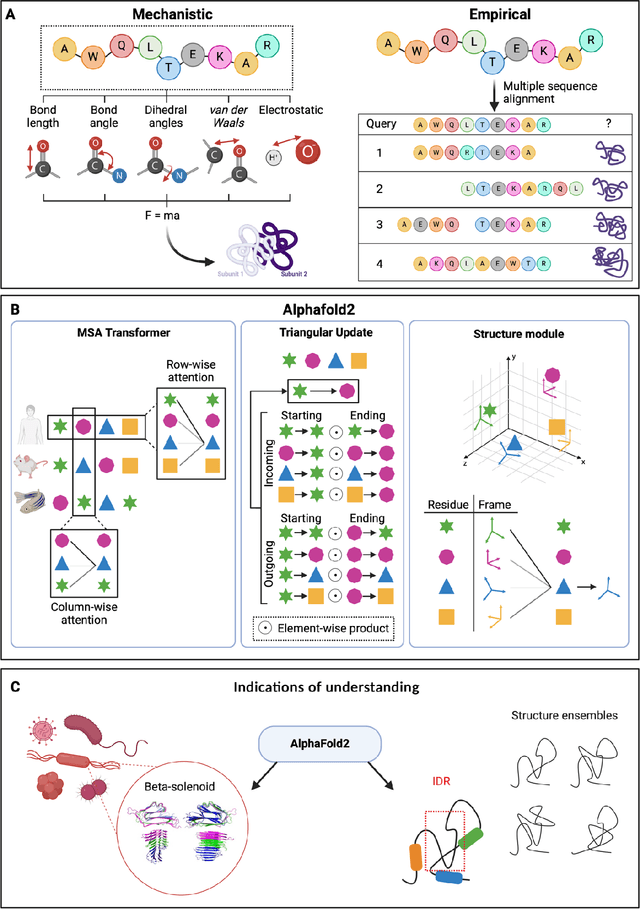
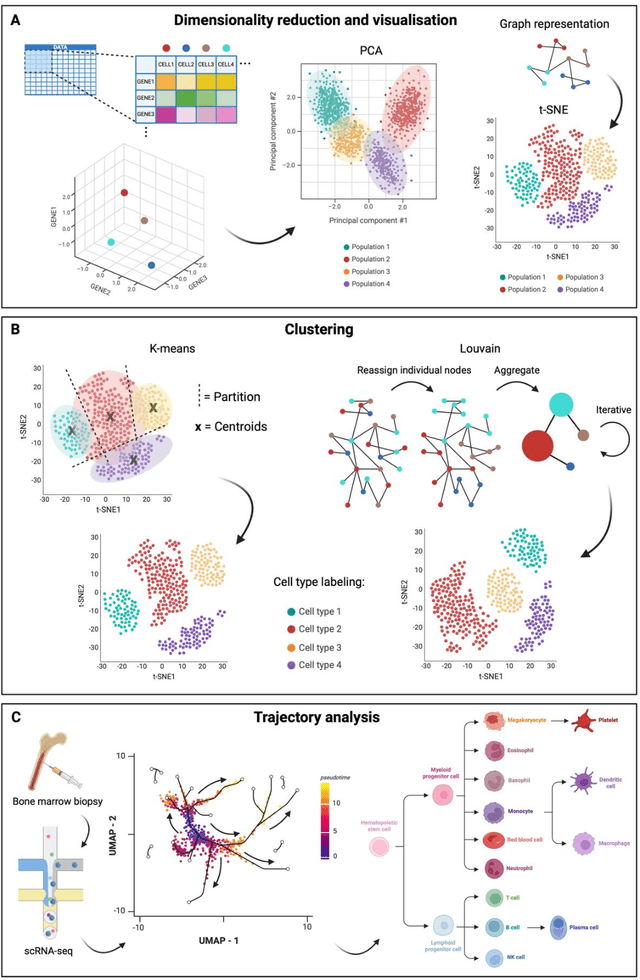
Modern life sciences research is increasingly relying on artificial intelligence approaches to model biological systems, primarily centered around the use of machine learning (ML) models. Although ML is undeniably useful for identifying patterns in large, complex data sets, its widespread application in biological sciences represents a significant deviation from traditional methods of scientific inquiry. As such, the interplay between these models and scientific understanding in biology is a topic with important implications for the future of scientific research, yet it is a subject that has received little attention. Here, we draw from an epistemological toolkit to contextualize recent applications of ML in biological sciences under modern philosophical theories of understanding, identifying general principles that can guide the design and application of ML systems to model biological phenomena and advance scientific knowledge. We propose that conceptions of scientific understanding as information compression, qualitative intelligibility, and dependency relation modelling provide a useful framework for interpreting ML-mediated understanding of biological systems. Through a detailed analysis of two key application areas of ML in modern biological research - protein structure prediction and single cell RNA-sequencing - we explore how these features have thus far enabled ML systems to advance scientific understanding of their target phenomena, how they may guide the development of future ML models, and the key obstacles that remain in preventing ML from achieving its potential as a tool for biological discovery. Consideration of the epistemological features of ML applications in biology will improve the prospects of these methods to solve important problems and advance scientific understanding of living systems.
Improving Antibody Humanness Prediction using Patent Data
Jan 31, 2024



We investigate the potential of patent data for improving the antibody humanness prediction using a multi-stage, multi-loss training process. Humanness serves as a proxy for the immunogenic response to antibody therapeutics, one of the major causes of attrition in drug discovery and a challenging obstacle for their use in clinical settings. We pose the initial learning stage as a weakly-supervised contrastive-learning problem, where each antibody sequence is associated with possibly multiple identifiers of function and the objective is to learn an encoder that groups them according to their patented properties. We then freeze a part of the contrastive encoder and continue training it on the patent data using the cross-entropy loss to predict the humanness score of a given antibody sequence. We illustrate the utility of the patent data and our approach by performing inference on three different immunogenicity datasets, unseen during training. Our empirical results demonstrate that the learned model consistently outperforms the alternative baselines and establishes new state-of-the-art on five out of six inference tasks, irrespective of the used metric.
Attentive cross-modal paratope prediction
Jun 12, 2018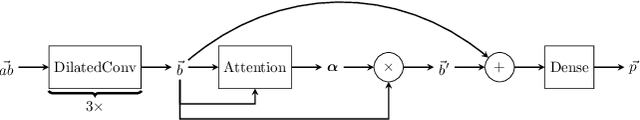

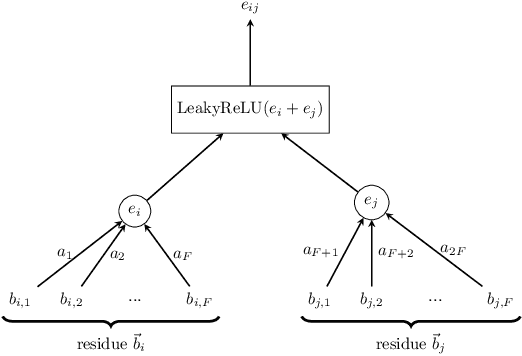
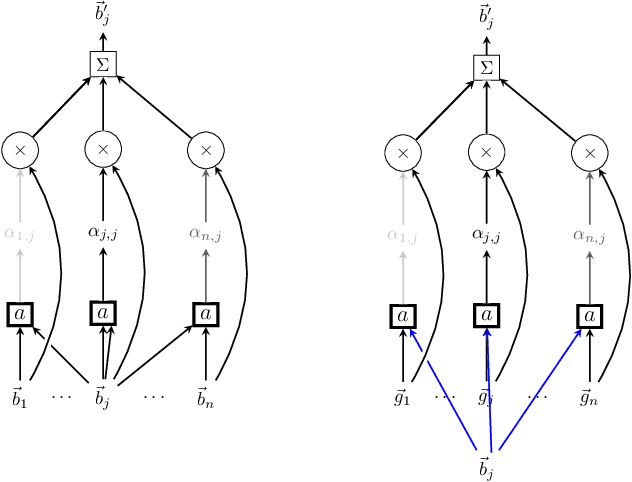
Antibodies are a critical part of the immune system, having the function of directly neutralising or tagging undesirable objects (the antigens) for future destruction. Being able to predict which amino acids belong to the paratope, the region on the antibody which binds to the antigen, can facilitate antibody design and contribute to the development of personalised medicine. The suitability of deep neural networks has recently been confirmed for this task, with Parapred outperforming all prior physical models. Our contribution is twofold: first, we significantly outperform the computational efficiency of Parapred by leveraging \`a trous convolutions and self-attention. Secondly, we implement cross-modal attention by allowing the antibody residues to attend over antigen residues. This leads to new state-of-the-art results on this task, along with insightful interpretations.