 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGeneral Multimodal Protein Design Enables DNA-Encoding of Chemistry
Apr 06, 2026Evolution is an extraordinary engine for enzymatic diversity, yet the chemistry it has explored remains a narrow slice of what DNA can encode. Deep generative models can design new proteins that bind ligands, but none have created enzymes without pre-specifying catalytic residues. We introduce DISCO (DIffusion for Sequence-structure CO-design), a multimodal model that co-designs protein sequence and 3D structure around arbitrary biomolecules, as well as inference-time scaling methods that optimize objectives across both modalities. Conditioned solely on reactive intermediates, DISCO designs diverse heme enzymes with novel active-site geometries. These enzymes catalyze new-to-nature carbene-transfer reactions, including alkene cyclopropanation, spirocyclopropanation, B-H, and C(sp$^3$)-H insertions, with high activities exceeding those of engineered enzymes. Random mutagenesis of a selected design further confirmed that enzyme activity can be improved through directed evolution. By providing a scalable route to evolvable enzymes, DISCO broadens the potential scope of genetically encodable transformations. Code is available at https://github.com/DISCO-design/DISCO.
Riemannian MeanFlow
Feb 08, 2026Diffusion and flow models have become the dominant paradigm for generative modeling on Riemannian manifolds, with successful applications in protein backbone generation and DNA sequence design. However, these methods require tens to hundreds of neural network evaluations at inference time, which can become a computational bottleneck in large-scale scientific sampling workflows. We introduce Riemannian MeanFlow~(RMF), a framework for learning flow maps directly on manifolds, enabling high-quality generations with as few as one forward pass. We derive three equivalent characterizations of the manifold average velocity (Eulerian, Lagrangian, and semigroup identities), and analyze parameterizations and stabilization techniques to improve training on high-dimensional manifolds. In promoter DNA design and protein backbone generation settings, RMF achieves comparable sample quality to prior methods while requiring up to 10$\times$ fewer function evaluations. Finally, we show that few-step flow maps enable efficient reward-guided design through reward look-ahead, where terminal states can be predicted from intermediate steps at minimal additional cost.
Discrete Feynman-Kac Correctors
Jan 15, 2026Discrete diffusion models have recently emerged as a promising alternative to the autoregressive approach for generating discrete sequences. Sample generation via gradual denoising or demasking processes allows them to capture hierarchical non-sequential interdependencies in the data. These custom processes, however, do not assume a flexible control over the distribution of generated samples. We propose Discrete Feynman-Kac Correctors, a framework that allows for controlling the generated distribution of discrete masked diffusion models at inference time. We derive Sequential Monte Carlo (SMC) algorithms that, given a trained discrete diffusion model, control the temperature of the sampled distribution (i.e. perform annealing), sample from the product of marginals of several diffusion processes (e.g. differently conditioned processes), and sample from the product of the marginal with an external reward function, producing likely samples from the target distribution that also have high reward. Notably, our framework does not require any training of additional models or fine-tuning of the original model. We illustrate the utility of our framework in several applications including: efficient sampling from the annealed Boltzmann distribution of the Ising model, improving the performance of language models for code generation and amortized learning, as well as reward-tilted protein sequence generation.
Amortized Sampling with Transferable Normalizing Flows
Aug 25, 2025Efficient equilibrium sampling of molecular conformations remains a core challenge in computational chemistry and statistical inference. Classical approaches such as molecular dynamics or Markov chain Monte Carlo inherently lack amortization; the computational cost of sampling must be paid in-full for each system of interest. The widespread success of generative models has inspired interest into overcoming this limitation through learning sampling algorithms. Despite performing on par with conventional methods when trained on a single system, learned samplers have so far demonstrated limited ability to transfer across systems. We prove that deep learning enables the design of scalable and transferable samplers by introducing Prose, a 280 million parameter all-atom transferable normalizing flow trained on a corpus of peptide molecular dynamics trajectories up to 8 residues in length. Prose draws zero-shot uncorrelated proposal samples for arbitrary peptide systems, achieving the previously intractable transferability across sequence length, whilst retaining the efficient likelihood evaluation of normalizing flows. Through extensive empirical evaluation we demonstrate the efficacy of Prose as a proposal for a variety of sampling algorithms, finding a simple importance sampling-based finetuning procedure to achieve superior performance to established methods such as sequential Monte Carlo on unseen tetrapeptides. We open-source the Prose codebase, model weights, and training dataset, to further stimulate research into amortized sampling methods and finetuning objectives.
Feynman-Kac Correctors in Diffusion: Annealing, Guidance, and Product of Experts
Mar 04, 2025While score-based generative models are the model of choice across diverse domains, there are limited tools available for controlling inference-time behavior in a principled manner, e.g. for composing multiple pretrained models. Existing classifier-free guidance methods use a simple heuristic to mix conditional and unconditional scores to approximately sample from conditional distributions. However, such methods do not approximate the intermediate distributions, necessitating additional 'corrector' steps. In this work, we provide an efficient and principled method for sampling from a sequence of annealed, geometric-averaged, or product distributions derived from pretrained score-based models. We derive a weighted simulation scheme which we call Feynman-Kac Correctors (FKCs) based on the celebrated Feynman-Kac formula by carefully accounting for terms in the appropriate partial differential equations (PDEs). To simulate these PDEs, we propose Sequential Monte Carlo (SMC) resampling algorithms that leverage inference-time scaling to improve sampling quality. We empirically demonstrate the utility of our methods by proposing amortized sampling via inference-time temperature annealing, improving multi-objective molecule generation using pretrained models, and improving classifier-free guidance for text-to-image generation. Our code is available at https://github.com/martaskrt/fkc-diffusion.
The Superposition of Diffusion Models Using the Itô Density Estimator
Dec 23, 2024The Cambrian explosion of easily accessible pre-trained diffusion models suggests a demand for methods that combine multiple different pre-trained diffusion models without incurring the significant computational burden of re-training a larger combined model. In this paper, we cast the problem of combining multiple pre-trained diffusion models at the generation stage under a novel proposed framework termed superposition. Theoretically, we derive superposition from rigorous first principles stemming from the celebrated continuity equation and design two novel algorithms tailor-made for combining diffusion models in SuperDiff. SuperDiff leverages a new scalable It\^o density estimator for the log likelihood of the diffusion SDE which incurs no additional overhead compared to the well-known Hutchinson's estimator needed for divergence calculations. We demonstrate that SuperDiff is scalable to large pre-trained diffusion models as superposition is performed solely through composition during inference, and also enjoys painless implementation as it combines different pre-trained vector fields through an automated re-weighting scheme. Notably, we show that SuperDiff is efficient during inference time, and mimics traditional composition operators such as the logical OR and the logical AND. We empirically demonstrate the utility of using SuperDiff for generating more diverse images on CIFAR-10, more faithful prompt conditioned image editing using Stable Diffusion, and improved unconditional de novo structure design of proteins. https://github.com/necludov/super-diffusion
Doob's Lagrangian: A Sample-Efficient Variational Approach to Transition Path Sampling
Oct 10, 2024

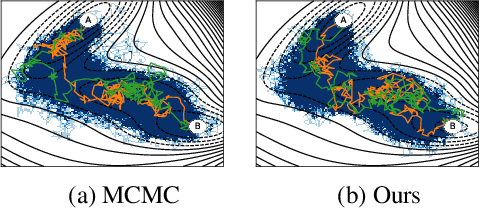
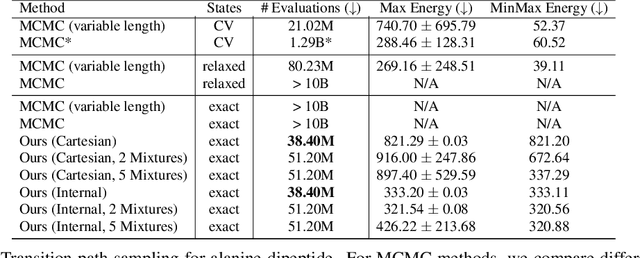
Rare event sampling in dynamical systems is a fundamental problem arising in the natural sciences, which poses significant computational challenges due to an exponentially large space of trajectories. For settings where the dynamical system of interest follows a Brownian motion with known drift, the question of conditioning the process to reach a given endpoint or desired rare event is definitively answered by Doob's h-transform. However, the naive estimation of this transform is infeasible, as it requires simulating sufficiently many forward trajectories to estimate rare event probabilities. In this work, we propose a variational formulation of Doob's $h$-transform as an optimization problem over trajectories between a given initial point and the desired ending point. To solve this optimization, we propose a simulation-free training objective with a model parameterization that imposes the desired boundary conditions by design. Our approach significantly reduces the search space over trajectories and avoids expensive trajectory simulation and inefficient importance sampling estimators which are required in existing methods. We demonstrate the ability of our method to find feasible transition paths on real-world molecular simulation and protein folding tasks.
Meta Flow Matching: Integrating Vector Fields on the Wasserstein Manifold
Aug 26, 2024



Numerous biological and physical processes can be modeled as systems of interacting entities evolving continuously over time, e.g. the dynamics of communicating cells or physical particles. Learning the dynamics of such systems is essential for predicting the temporal evolution of populations across novel samples and unseen environments. Flow-based models allow for learning these dynamics at the population level - they model the evolution of the entire distribution of samples. However, current flow-based models are limited to a single initial population and a set of predefined conditions which describe different dynamics. We argue that multiple processes in natural sciences have to be represented as vector fields on the Wasserstein manifold of probability densities. That is, the change of the population at any moment in time depends on the population itself due to the interactions between samples. In particular, this is crucial for personalized medicine where the development of diseases and their respective treatment response depends on the microenvironment of cells specific to each patient. We propose Meta Flow Matching (MFM), a practical approach to integrating along these vector fields on the Wasserstein manifold by amortizing the flow model over the initial populations. Namely, we embed the population of samples using a Graph Neural Network (GNN) and use these embeddings to train a Flow Matching model. This gives MFM the ability to generalize over the initial distributions unlike previously proposed methods. We demonstrate the ability of MFM to improve prediction of individual treatment responses on a large scale multi-patient single-cell drug screen dataset.
Efficient Evolutionary Search Over Chemical Space with Large Language Models
Jun 23, 2024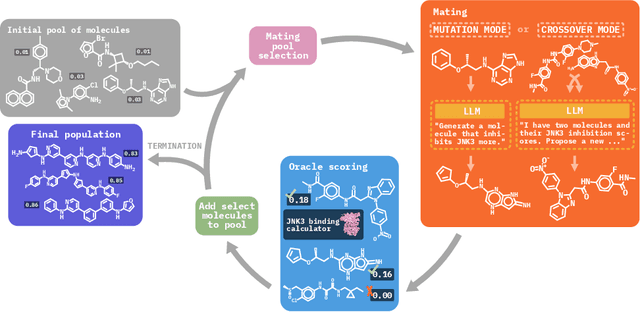
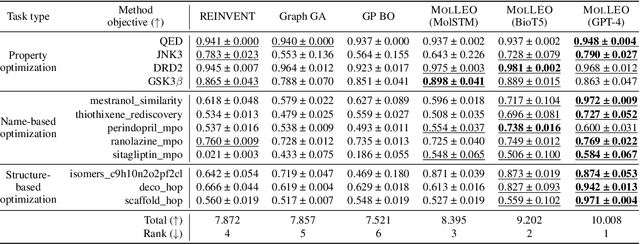
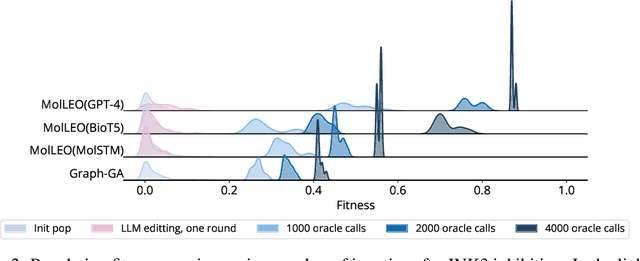
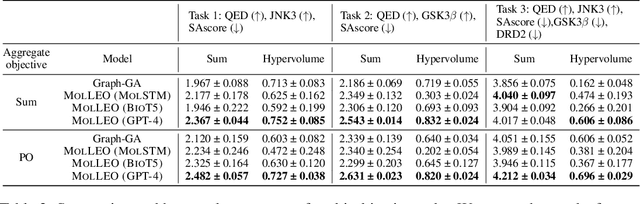
Molecular discovery, when formulated as an optimization problem, presents significant computational challenges because optimization objectives can be non-differentiable. Evolutionary Algorithms (EAs), often used to optimize black-box objectives in molecular discovery, traverse chemical space by performing random mutations and crossovers, leading to a large number of expensive objective evaluations. In this work, we ameliorate this shortcoming by incorporating chemistry-aware Large Language Models (LLMs) into EAs. Namely, we redesign crossover and mutation operations in EAs using LLMs trained on large corpora of chemical information. We perform extensive empirical studies on both commercial and open-source models on multiple tasks involving property optimization, molecular rediscovery, and structure-based drug design, demonstrating that the joint usage of LLMs with EAs yields superior performance over all baseline models across single- and multi-objective settings. We demonstrate that our algorithm improves both the quality of the final solution and convergence speed, thereby reducing the number of required objective evaluations. Our code is available at http://github.com/zoom-wang112358/MOLLEO
Diffusion Models as Constrained Samplers for Optimization with Unknown Constraints
Feb 28, 2024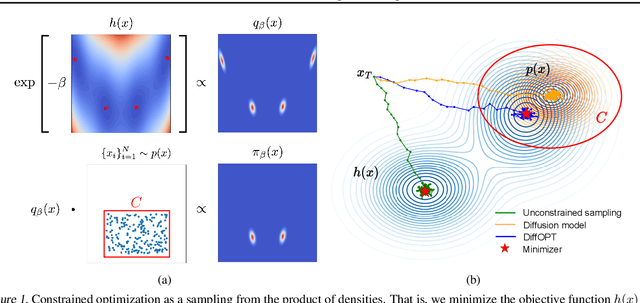
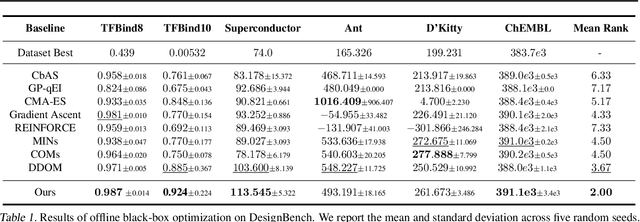
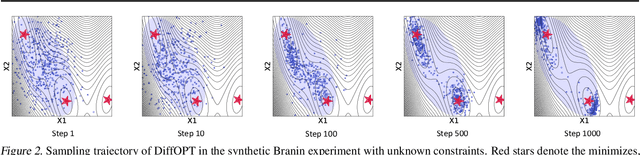
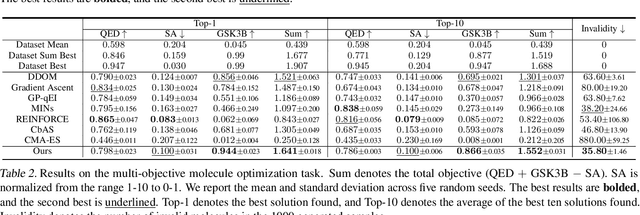
Addressing real-world optimization problems becomes particularly challenging when analytic objective functions or constraints are unavailable. While numerous studies have addressed the issue of unknown objectives, limited research has focused on scenarios where feasibility constraints are not given explicitly. Overlooking these constraints can lead to spurious solutions that are unrealistic in practice. To deal with such unknown constraints, we propose to perform optimization within the data manifold using diffusion models. To constrain the optimization process to the data manifold, we reformulate the original optimization problem as a sampling problem from the product of the Boltzmann distribution defined by the objective function and the data distribution learned by the diffusion model. To enhance sampling efficiency, we propose a two-stage framework that begins with a guided diffusion process for warm-up, followed by a Langevin dynamics stage for further correction. Theoretical analysis shows that the initial stage results in a distribution focused on feasible solutions, thereby providing a better initialization for the later stage. Comprehensive experiments on a synthetic dataset, six real-world black-box optimization datasets, and a multi-objective optimization dataset show that our method achieves better or comparable performance with previous state-of-the-art baselines.