 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFALCON: Few-step Accurate Likelihoods for Continuous Flows
Dec 10, 2025

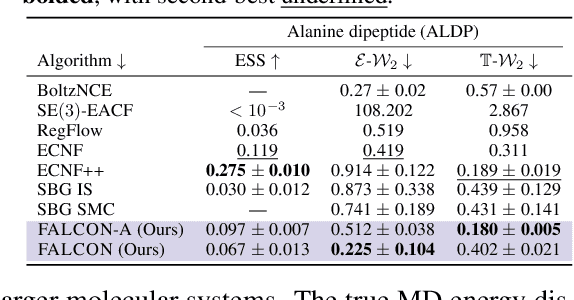

Scalable sampling of molecular states in thermodynamic equilibrium is a long-standing challenge in statistical physics. Boltzmann Generators tackle this problem by pairing a generative model, capable of exact likelihood computation, with importance sampling to obtain consistent samples under the target distribution. Current Boltzmann Generators primarily use continuous normalizing flows (CNFs) trained with flow matching for efficient training of powerful models. However, likelihood calculation for these models is extremely costly, requiring thousands of function evaluations per sample, severely limiting their adoption. In this work, we propose Few-step Accurate Likelihoods for Continuous Flows (FALCON), a method which allows for few-step sampling with a likelihood accurate enough for importance sampling applications by introducing a hybrid training objective that encourages invertibility. We show FALCON outperforms state-of-the-art normalizing flow models for molecular Boltzmann sampling and is two orders of magnitude faster than the equivalently performing CNF model.
Feynman-Kac Correctors in Diffusion: Annealing, Guidance, and Product of Experts
Mar 04, 2025While score-based generative models are the model of choice across diverse domains, there are limited tools available for controlling inference-time behavior in a principled manner, e.g. for composing multiple pretrained models. Existing classifier-free guidance methods use a simple heuristic to mix conditional and unconditional scores to approximately sample from conditional distributions. However, such methods do not approximate the intermediate distributions, necessitating additional 'corrector' steps. In this work, we provide an efficient and principled method for sampling from a sequence of annealed, geometric-averaged, or product distributions derived from pretrained score-based models. We derive a weighted simulation scheme which we call Feynman-Kac Correctors (FKCs) based on the celebrated Feynman-Kac formula by carefully accounting for terms in the appropriate partial differential equations (PDEs). To simulate these PDEs, we propose Sequential Monte Carlo (SMC) resampling algorithms that leverage inference-time scaling to improve sampling quality. We empirically demonstrate the utility of our methods by proposing amortized sampling via inference-time temperature annealing, improving multi-objective molecule generation using pretrained models, and improving classifier-free guidance for text-to-image generation. Our code is available at https://github.com/martaskrt/fkc-diffusion.
Symmetry-Aware Generative Modeling through Learned Canonicalization
Jan 14, 2025Generative modeling of symmetric densities has a range of applications in AI for science, from drug discovery to physics simulations. The existing generative modeling paradigm for invariant densities combines an invariant prior with an equivariant generative process. However, we observe that this technique is not necessary and has several drawbacks resulting from the limitations of equivariant networks. Instead, we propose to model a learned slice of the density so that only one representative element per orbit is learned. To accomplish this, we learn a group-equivariant canonicalization network that maps training samples to a canonical pose and train a non-equivariant generative model over these canonicalized samples. We implement this idea in the context of diffusion models. Our preliminary experimental results on molecular modeling are promising, demonstrating improved sample quality and faster inference time.
Sampling from Energy-based Policies using Diffusion
Oct 02, 2024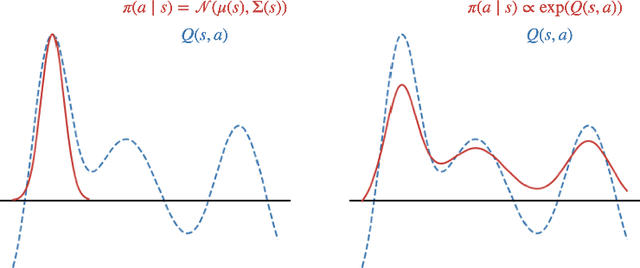
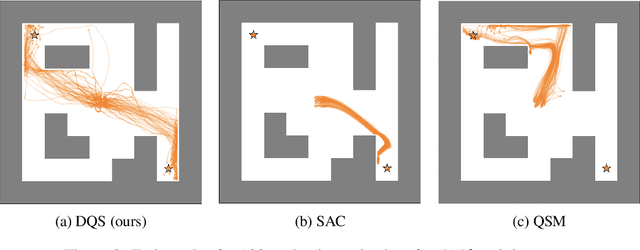

Energy-based policies offer a flexible framework for modeling complex, multimodal behaviors in reinforcement learning (RL). In maximum entropy RL, the optimal policy is a Boltzmann distribution derived from the soft Q-function, but direct sampling from this distribution in continuous action spaces is computationally intractable. As a result, existing methods typically use simpler parametric distributions, like Gaussians, for policy representation - limiting their ability to capture the full complexity of multimodal action distributions. In this paper, we introduce a diffusion-based approach for sampling from energy-based policies, where the negative Q-function defines the energy function. Based on this approach, we propose an actor-critic method called Diffusion Q-Sampling (DQS) that enables more expressive policy representations, allowing stable learning in diverse environments. We show that our approach enhances exploration and captures multimodal behavior in continuous control tasks, addressing key limitations of existing methods.
Sequence-Augmented SE(3)-Flow Matching For Conditional Protein Backbone Generation
May 30, 2024Proteins are essential for almost all biological processes and derive their diverse functions from complex 3D structures, which are in turn determined by their amino acid sequences. In this paper, we exploit the rich biological inductive bias of amino acid sequences and introduce FoldFlow-2, a novel sequence-conditioned SE(3)-equivariant flow matching model for protein structure generation. FoldFlow-2 presents substantial new architectural features over the previous FoldFlow family of models including a protein large language model to encode sequence, a new multi-modal fusion trunk that combines structure and sequence representations, and a geometric transformer based decoder. To increase diversity and novelty of generated samples -- crucial for de-novo drug design -- we train FoldFlow-2 at scale on a new dataset that is an order of magnitude larger than PDB datasets of prior works, containing both known proteins in PDB and high-quality synthetic structures achieved through filtering. We further demonstrate the ability to align FoldFlow-2 to arbitrary rewards, e.g. increasing secondary structures diversity, by introducing a Reinforced Finetuning (ReFT) objective. We empirically observe that FoldFlow-2 outperforms previous state-of-the-art protein structure-based generative models, improving over RFDiffusion in terms of unconditional generation across all metrics including designability, diversity, and novelty across all protein lengths, as well as exhibiting generalization on the task of equilibrium conformation sampling. Finally, we demonstrate that a fine-tuned FoldFlow-2 makes progress on challenging conditional design tasks such as designing scaffolds for the VHH nanobody.
Iterated Denoising Energy Matching for Sampling from Boltzmann Densities
Feb 09, 2024



Efficiently generating statistically independent samples from an unnormalized probability distribution, such as equilibrium samples of many-body systems, is a foundational problem in science. In this paper, we propose Iterated Denoising Energy Matching (iDEM), an iterative algorithm that uses a novel stochastic score matching objective leveraging solely the energy function and its gradient -- and no data samples -- to train a diffusion-based sampler. Specifically, iDEM alternates between (I) sampling regions of high model density from a diffusion-based sampler and (II) using these samples in our stochastic matching objective to further improve the sampler. iDEM is scalable to high dimensions as the inner matching objective, is simulation-free, and requires no MCMC samples. Moreover, by leveraging the fast mode mixing behavior of diffusion, iDEM smooths out the energy landscape enabling efficient exploration and learning of an amortized sampler. We evaluate iDEM on a suite of tasks ranging from standard synthetic energy functions to invariant $n$-body particle systems. We show that the proposed approach achieves state-of-the-art performance on all metrics and trains $2-5\times$ faster, which allows it to be the first method to train using energy on the challenging $55$-particle Lennard-Jones system.
Lie Point Symmetry and Physics Informed Networks
Nov 07, 2023



Symmetries have been leveraged to improve the generalization of neural networks through different mechanisms from data augmentation to equivariant architectures. However, despite their potential, their integration into neural solvers for partial differential equations (PDEs) remains largely unexplored. We explore the integration of PDE symmetries, known as Lie point symmetries, in a major family of neural solvers known as physics-informed neural networks (PINNs). We propose a loss function that informs the network about Lie point symmetries in the same way that PINN models try to enforce the underlying PDE through a loss function. Intuitively, our symmetry loss ensures that the infinitesimal generators of the Lie group conserve the PDE solutions. Effectively, this means that once the network learns a solution, it also learns the neighbouring solutions generated by Lie point symmetries. Empirical evaluations indicate that the inductive bias introduced by the Lie point symmetries of the PDEs greatly boosts the sample efficiency of PINNs.
SE(3)-Stochastic Flow Matching for Protein Backbone Generation
Oct 03, 2023The computational design of novel protein structures has the potential to impact numerous scientific disciplines greatly. Toward this goal, we introduce $\text{FoldFlow}$ a series of novel generative models of increasing modeling power based on the flow-matching paradigm over $3\text{D}$ rigid motions -- i.e. the group $\text{SE(3)}$ -- enabling accurate modeling of protein backbones. We first introduce $\text{FoldFlow-Base}$, a simulation-free approach to learning deterministic continuous-time dynamics and matching invariant target distributions on $\text{SE(3)}$. We next accelerate training by incorporating Riemannian optimal transport to create $\text{FoldFlow-OT}$, leading to the construction of both more simple and stable flows. Finally, we design $\text{FoldFlow-SFM}$ coupling both Riemannian OT and simulation-free training to learn stochastic continuous-time dynamics over $\text{SE(3)}$. Our family of $\text{FoldFlow}$ generative models offer several key advantages over previous approaches to the generative modeling of proteins: they are more stable and faster to train than diffusion-based approaches, and our models enjoy the ability to map any invariant source distribution to any invariant target distribution over $\text{SE(3)}$. Empirically, we validate our FoldFlow models on protein backbone generation of up to $300$ amino acids leading to high-quality designable, diverse, and novel samples.