 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGeneral Multimodal Protein Design Enables DNA-Encoding of Chemistry
Apr 06, 2026Evolution is an extraordinary engine for enzymatic diversity, yet the chemistry it has explored remains a narrow slice of what DNA can encode. Deep generative models can design new proteins that bind ligands, but none have created enzymes without pre-specifying catalytic residues. We introduce DISCO (DIffusion for Sequence-structure CO-design), a multimodal model that co-designs protein sequence and 3D structure around arbitrary biomolecules, as well as inference-time scaling methods that optimize objectives across both modalities. Conditioned solely on reactive intermediates, DISCO designs diverse heme enzymes with novel active-site geometries. These enzymes catalyze new-to-nature carbene-transfer reactions, including alkene cyclopropanation, spirocyclopropanation, B-H, and C(sp$^3$)-H insertions, with high activities exceeding those of engineered enzymes. Random mutagenesis of a selected design further confirmed that enzyme activity can be improved through directed evolution. By providing a scalable route to evolvable enzymes, DISCO broadens the potential scope of genetically encodable transformations. Code is available at https://github.com/DISCO-design/DISCO.
Riemannian MeanFlow
Feb 08, 2026Diffusion and flow models have become the dominant paradigm for generative modeling on Riemannian manifolds, with successful applications in protein backbone generation and DNA sequence design. However, these methods require tens to hundreds of neural network evaluations at inference time, which can become a computational bottleneck in large-scale scientific sampling workflows. We introduce Riemannian MeanFlow~(RMF), a framework for learning flow maps directly on manifolds, enabling high-quality generations with as few as one forward pass. We derive three equivalent characterizations of the manifold average velocity (Eulerian, Lagrangian, and semigroup identities), and analyze parameterizations and stabilization techniques to improve training on high-dimensional manifolds. In promoter DNA design and protein backbone generation settings, RMF achieves comparable sample quality to prior methods while requiring up to 10$\times$ fewer function evaluations. Finally, we show that few-step flow maps enable efficient reward-guided design through reward look-ahead, where terminal states can be predicted from intermediate steps at minimal additional cost.
Discrete Feynman-Kac Correctors
Jan 15, 2026Discrete diffusion models have recently emerged as a promising alternative to the autoregressive approach for generating discrete sequences. Sample generation via gradual denoising or demasking processes allows them to capture hierarchical non-sequential interdependencies in the data. These custom processes, however, do not assume a flexible control over the distribution of generated samples. We propose Discrete Feynman-Kac Correctors, a framework that allows for controlling the generated distribution of discrete masked diffusion models at inference time. We derive Sequential Monte Carlo (SMC) algorithms that, given a trained discrete diffusion model, control the temperature of the sampled distribution (i.e. perform annealing), sample from the product of marginals of several diffusion processes (e.g. differently conditioned processes), and sample from the product of the marginal with an external reward function, producing likely samples from the target distribution that also have high reward. Notably, our framework does not require any training of additional models or fine-tuning of the original model. We illustrate the utility of our framework in several applications including: efficient sampling from the annealed Boltzmann distribution of the Ising model, improving the performance of language models for code generation and amortized learning, as well as reward-tilted protein sequence generation.
Feynman-Kac Correctors in Diffusion: Annealing, Guidance, and Product of Experts
Mar 04, 2025While score-based generative models are the model of choice across diverse domains, there are limited tools available for controlling inference-time behavior in a principled manner, e.g. for composing multiple pretrained models. Existing classifier-free guidance methods use a simple heuristic to mix conditional and unconditional scores to approximately sample from conditional distributions. However, such methods do not approximate the intermediate distributions, necessitating additional 'corrector' steps. In this work, we provide an efficient and principled method for sampling from a sequence of annealed, geometric-averaged, or product distributions derived from pretrained score-based models. We derive a weighted simulation scheme which we call Feynman-Kac Correctors (FKCs) based on the celebrated Feynman-Kac formula by carefully accounting for terms in the appropriate partial differential equations (PDEs). To simulate these PDEs, we propose Sequential Monte Carlo (SMC) resampling algorithms that leverage inference-time scaling to improve sampling quality. We empirically demonstrate the utility of our methods by proposing amortized sampling via inference-time temperature annealing, improving multi-objective molecule generation using pretrained models, and improving classifier-free guidance for text-to-image generation. Our code is available at https://github.com/martaskrt/fkc-diffusion.
The Superposition of Diffusion Models Using the Itô Density Estimator
Dec 23, 2024The Cambrian explosion of easily accessible pre-trained diffusion models suggests a demand for methods that combine multiple different pre-trained diffusion models without incurring the significant computational burden of re-training a larger combined model. In this paper, we cast the problem of combining multiple pre-trained diffusion models at the generation stage under a novel proposed framework termed superposition. Theoretically, we derive superposition from rigorous first principles stemming from the celebrated continuity equation and design two novel algorithms tailor-made for combining diffusion models in SuperDiff. SuperDiff leverages a new scalable It\^o density estimator for the log likelihood of the diffusion SDE which incurs no additional overhead compared to the well-known Hutchinson's estimator needed for divergence calculations. We demonstrate that SuperDiff is scalable to large pre-trained diffusion models as superposition is performed solely through composition during inference, and also enjoys painless implementation as it combines different pre-trained vector fields through an automated re-weighting scheme. Notably, we show that SuperDiff is efficient during inference time, and mimics traditional composition operators such as the logical OR and the logical AND. We empirically demonstrate the utility of using SuperDiff for generating more diverse images on CIFAR-10, more faithful prompt conditioned image editing using Stable Diffusion, and improved unconditional de novo structure design of proteins. https://github.com/necludov/super-diffusion
Quantum Deep Equilibrium Models
Oct 31, 2024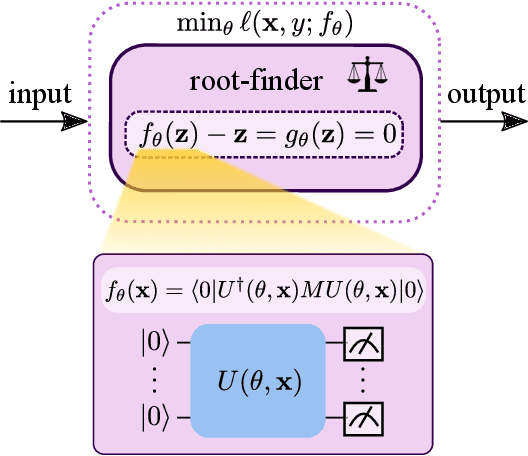
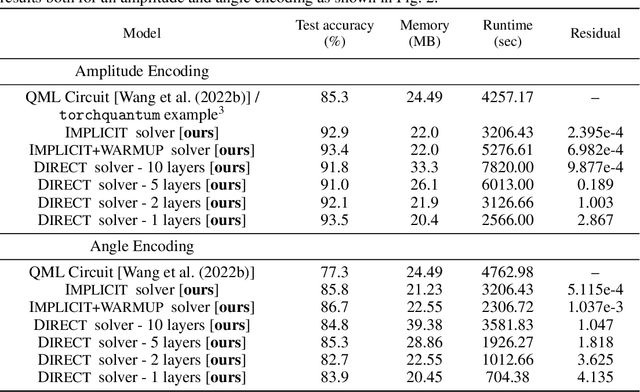
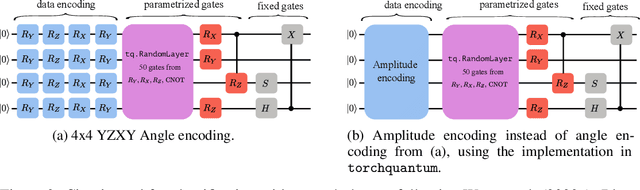
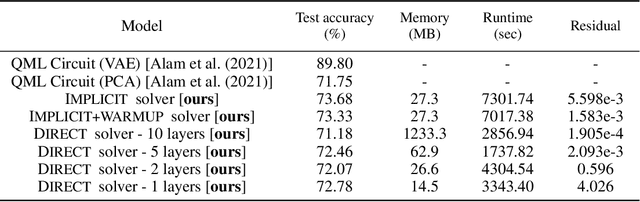
The feasibility of variational quantum algorithms, the most popular correspondent of neural networks on noisy, near-term quantum hardware, is highly impacted by the circuit depth of the involved parametrized quantum circuits (PQCs). Higher depth increases expressivity, but also results in a detrimental accumulation of errors. Furthermore, the number of parameters involved in the PQC significantly influences the performance through the necessary number of measurements to evaluate gradients, which scales linearly with the number of parameters. Motivated by this, we look at deep equilibrium models (DEQs), which mimic an infinite-depth, weight-tied network using a fraction of the memory by employing a root solver to find the fixed points of the network. In this work, we present Quantum Deep Equilibrium Models (QDEQs): a training paradigm that learns parameters of a quantum machine learning model given by a PQC using DEQs. To our knowledge, no work has yet explored the application of DEQs to QML models. We apply QDEQs to find the parameters of a quantum circuit in two settings: the first involves classifying MNIST-4 digits with 4 qubits; the second extends it to 10 classes of MNIST, FashionMNIST and CIFAR. We find that QDEQ is not only competitive with comparable existing baseline models, but also achieves higher performance than a network with 5 times more layers. This demonstrates that the QDEQ paradigm can be used to develop significantly more shallow quantum circuits for a given task, something which is essential for the utility of near-term quantum computers. Our code is available at https://github.com/martaskrt/qdeq.
How to do impactful research in artificial intelligence for chemistry and materials science
Sep 16, 2024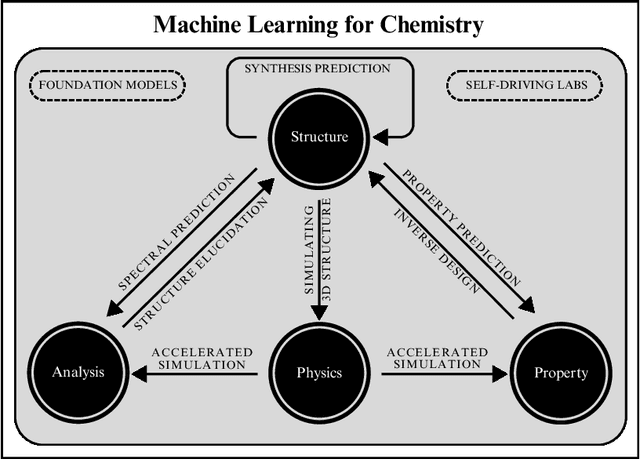
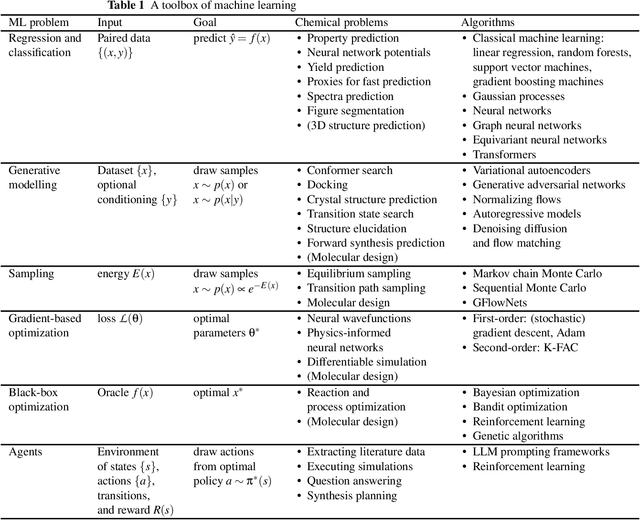
Machine learning has been pervasively touching many fields of science. Chemistry and materials science are no exception. While machine learning has been making a great impact, it is still not reaching its full potential or maturity. In this perspective, we first outline current applications across a diversity of problems in chemistry. Then, we discuss how machine learning researchers view and approach problems in the field. Finally, we provide our considerations for maximizing impact when researching machine learning for chemistry.
Efficient Evolutionary Search Over Chemical Space with Large Language Models
Jun 23, 2024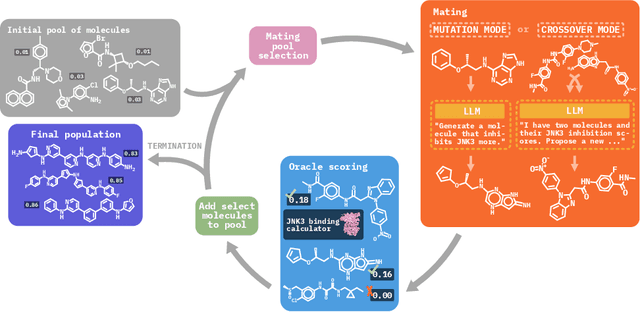
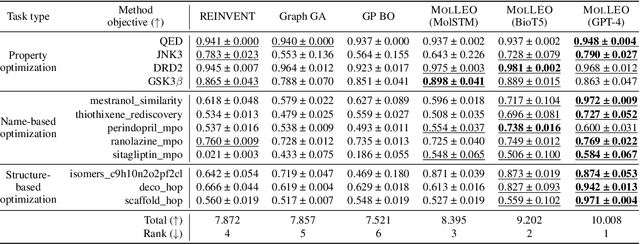
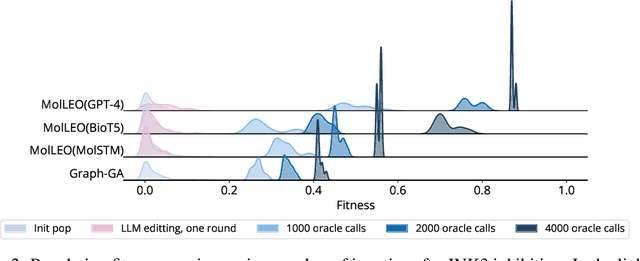
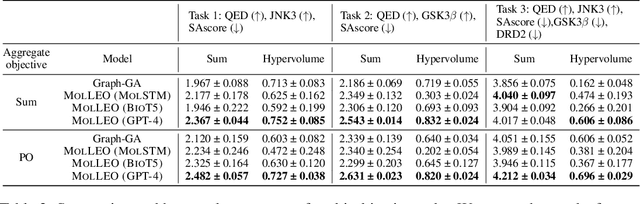
Molecular discovery, when formulated as an optimization problem, presents significant computational challenges because optimization objectives can be non-differentiable. Evolutionary Algorithms (EAs), often used to optimize black-box objectives in molecular discovery, traverse chemical space by performing random mutations and crossovers, leading to a large number of expensive objective evaluations. In this work, we ameliorate this shortcoming by incorporating chemistry-aware Large Language Models (LLMs) into EAs. Namely, we redesign crossover and mutation operations in EAs using LLMs trained on large corpora of chemical information. We perform extensive empirical studies on both commercial and open-source models on multiple tasks involving property optimization, molecular rediscovery, and structure-based drug design, demonstrating that the joint usage of LLMs with EAs yields superior performance over all baseline models across single- and multi-objective settings. We demonstrate that our algorithm improves both the quality of the final solution and convergence speed, thereby reducing the number of required objective evaluations. Our code is available at http://github.com/zoom-wang112358/MOLLEO
A Sober Look at LLMs for Material Discovery: Are They Actually Good for Bayesian Optimization Over Molecules?
Feb 07, 2024



Automation is one of the cornerstones of contemporary material discovery. Bayesian optimization (BO) is an essential part of such workflows, enabling scientists to leverage prior domain knowledge into efficient exploration of a large molecular space. While such prior knowledge can take many forms, there has been significant fanfare around the ancillary scientific knowledge encapsulated in large language models (LLMs). However, existing work thus far has only explored LLMs for heuristic materials searches. Indeed, recent work obtains the uncertainty estimate -- an integral part of BO -- from point-estimated, non-Bayesian LLMs. In this work, we study the question of whether LLMs are actually useful to accelerate principled Bayesian optimization in the molecular space. We take a sober, dispassionate stance in answering this question. This is done by carefully (i) viewing LLMs as fixed feature extractors for standard but principled BO surrogate models and by (ii) leveraging parameter-efficient finetuning methods and Bayesian neural networks to obtain the posterior of the LLM surrogate. Our extensive experiments with real-world chemistry problems show that LLMs can be useful for BO over molecules, but only if they have been pretrained or finetuned with domain-specific data.
ORGANA: A Robotic Assistant for Automated Chemistry Experimentation and Characterization
Jan 13, 2024



Chemistry experimentation is often resource- and labor-intensive. Despite the many benefits incurred by the integration of advanced and special-purpose lab equipment, many aspects of experimentation are still manually conducted by chemists, for example, polishing an electrode in electrochemistry experiments. Traditional lab automation infrastructure faces challenges when it comes to flexibly adapting to new chemistry experiments. To address this issue, we propose a human-friendly and flexible robotic system, ORGANA, that automates a diverse set of chemistry experiments. It is capable of interacting with chemists in the lab through natural language, using Large Language Models (LLMs). ORGANA keeps scientists informed by providing timely reports that incorporate statistical analyses. Additionally, it actively engages with users when necessary for disambiguation or troubleshooting. ORGANA can reason over user input to derive experiment goals, and plan long sequences of both high-level tasks and low-level robot actions while using feedback from the visual perception of the environment. It also supports scheduling and parallel execution for experiments that require resource allocation and coordination between multiple robots and experiment stations. We show that ORGANA successfully conducts a diverse set of chemistry experiments, including solubility assessment, pH measurement, recrystallization, and electrochemistry experiments. For the latter, we show that ORGANA robustly executes a long-horizon plan, comprising 19 steps executed in parallel, to characterize the electrochemical properties of quinone derivatives, a class of molecules used in rechargeable flow batteries. Our user study indicates that ORGANA significantly improves many aspects of user experience while reducing their physical workload. More details about ORGANA can be found at https://ac-rad.github.io/organa/.