 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
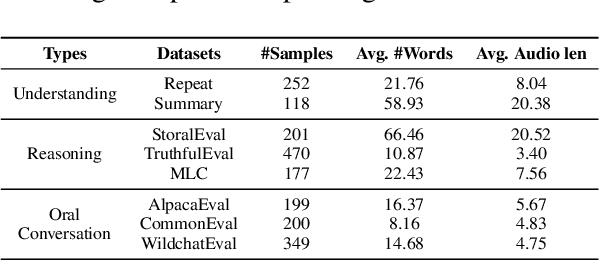
Add to EdgeSLAM-LLM: A Modular, Open-Source Multimodal Large Language Model Framework and Best Practice for Speech, Language, Audio and Music Processing
Jan 14, 2026The recent surge in open-source Multimodal Large Language Models (MLLM) frameworks, such as LLaVA, provides a convenient kickoff for artificial intelligence developers and researchers. However, most of the MLLM frameworks take vision as the main input modality, and provide limited in-depth support for the modality of speech, audio, and music. This situation hinders the development of audio-language models, and forces researchers to spend a lot of effort on code writing and hyperparameter tuning. We present SLAM-LLM, an open-source deep learning framework designed to train customized MLLMs, focused on speech, language, audio, and music processing. SLAM-LLM provides a modular configuration of different encoders, projectors, LLMs, and parameter-efficient fine-tuning plugins. SLAM-LLM also includes detailed training and inference recipes for mainstream tasks, along with high-performance checkpoints like LLM-based Automatic Speech Recognition (ASR), Automated Audio Captioning (AAC), and Music Captioning (MC). Some of these recipes have already reached or are nearing state-of-the-art performance, and some relevant techniques have also been accepted by academic papers. We hope SLAM-LLM will accelerate iteration, development, data engineering, and model training for researchers. We are committed to continually pushing forward audio-based MLLMs through this open-source framework, and call on the community to contribute to the LLM-based speech, audio and music processing.
MMAR: A Challenging Benchmark for Deep Reasoning in Speech, Audio, Music, and Their Mix
May 19, 2025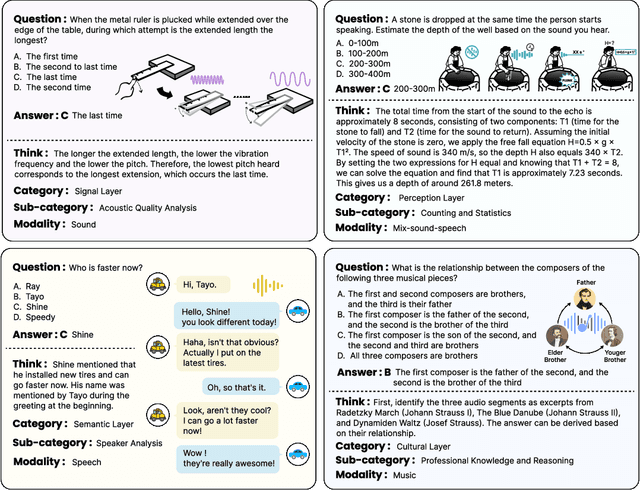

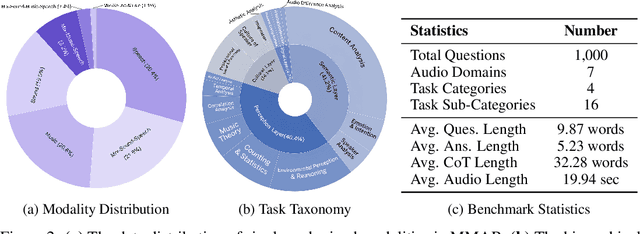
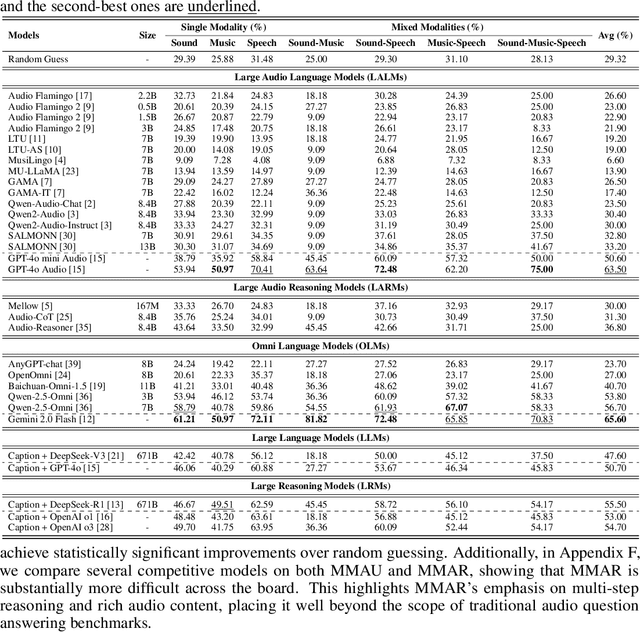
We introduce MMAR, a new benchmark designed to evaluate the deep reasoning capabilities of Audio-Language Models (ALMs) across massive multi-disciplinary tasks. MMAR comprises 1,000 meticulously curated audio-question-answer triplets, collected from real-world internet videos and refined through iterative error corrections and quality checks to ensure high quality. Unlike existing benchmarks that are limited to specific domains of sound, music, or speech, MMAR extends them to a broad spectrum of real-world audio scenarios, including mixed-modality combinations of sound, music, and speech. Each question in MMAR is hierarchically categorized across four reasoning layers: Signal, Perception, Semantic, and Cultural, with additional sub-categories within each layer to reflect task diversity and complexity. To further foster research in this area, we annotate every question with a Chain-of-Thought (CoT) rationale to promote future advancements in audio reasoning. Each item in the benchmark demands multi-step deep reasoning beyond surface-level understanding. Moreover, a part of the questions requires graduate-level perceptual and domain-specific knowledge, elevating the benchmark's difficulty and depth. We evaluate MMAR using a broad set of models, including Large Audio-Language Models (LALMs), Large Audio Reasoning Models (LARMs), Omni Language Models (OLMs), Large Language Models (LLMs), and Large Reasoning Models (LRMs), with audio caption inputs. The performance of these models on MMAR highlights the benchmark's challenging nature, and our analysis further reveals critical limitations of understanding and reasoning capabilities among current models. We hope MMAR will serve as a catalyst for future advances in this important but little-explored area.
Large Language Models Are Innate Crystal Structure Generators
Feb 28, 2025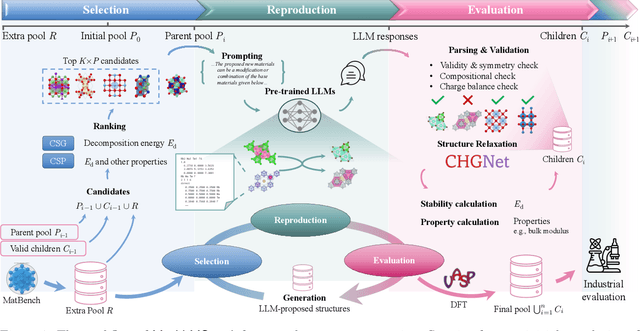
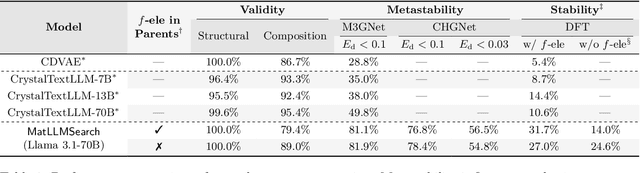
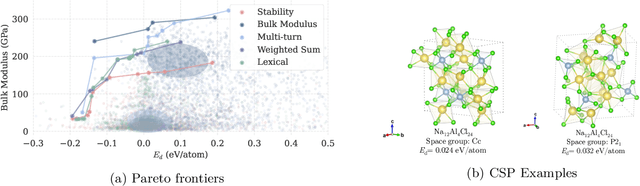
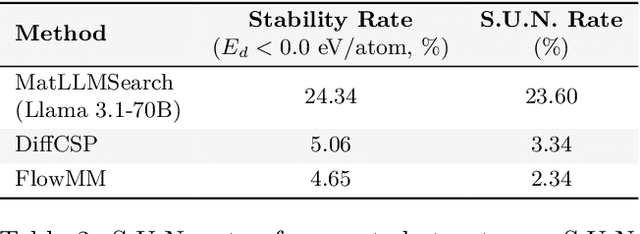
Crystal structure generation is fundamental to materials discovery, enabling the prediction of novel materials with desired properties. While existing approaches leverage Large Language Models (LLMs) through extensive fine-tuning on materials databases, we show that pre-trained LLMs can inherently generate stable crystal structures without additional training. Our novel framework MatLLMSearch integrates pre-trained LLMs with evolutionary search algorithms, achieving a 78.38% metastable rate validated by machine learning interatomic potentials and 31.7% DFT-verified stability via quantum mechanical calculations, outperforming specialized models such as CrystalTextLLM. Beyond crystal structure generation, we further demonstrate that our framework can be readily adapted to diverse materials design tasks, including crystal structure prediction and multi-objective optimization of properties such as deformation energy and bulk modulus, all without fine-tuning. These results establish pre-trained LLMs as versatile and effective tools for materials discovery, opening up new venues for crystal structure generation with reduced computational overhead and broader accessibility.
SLAM-Omni: Timbre-Controllable Voice Interaction System with Single-Stage Training
Dec 20, 2024

Recent advancements highlight the potential of end-to-end real-time spoken dialogue systems, showcasing their low latency and high quality. In this paper, we introduce SLAM-Omni, a timbre-controllable, end-to-end voice interaction system with single-stage training. SLAM-Omni achieves zero-shot timbre control by modeling spoken language with semantic tokens and decoupling speaker information to a vocoder. By predicting grouped speech semantic tokens at each step, our method significantly reduces the sequence length of audio tokens, accelerating both training and inference. Additionally, we propose historical text prompting to compress dialogue history, facilitating efficient multi-round interactions. Comprehensive evaluations reveal that SLAM-Omni outperforms prior models of similar scale, requiring only 15 hours of training on 4 GPUs with limited data. Notably, it is the first spoken dialogue system to achieve competitive performance with a single-stage training approach, eliminating the need for pre-training on TTS or ASR tasks. Further experiments validate its multilingual and multi-turn dialogue capabilities on larger datasets.
Bi-Level Graph Structure Learning for Next POI Recommendation
Nov 02, 2024



Next point-of-interest (POI) recommendation aims to predict a user's next destination based on sequential check-in history and a set of POI candidates. Graph neural networks (GNNs) have demonstrated a remarkable capability in this endeavor by exploiting the extensive global collaborative signals present among POIs. However, most of the existing graph-based approaches construct graph structures based on pre-defined heuristics, failing to consider inherent hierarchical structures of POI features such as geographical locations and visiting peaks, or suffering from noisy and incomplete structures in graphs. To address the aforementioned issues, this paper presents a novel Bi-level Graph Structure Learning (BiGSL) for next POI recommendation. BiGSL first learns a hierarchical graph structure to capture the fine-to-coarse connectivity between POIs and prototypes, and then uses a pairwise learning module to dynamically infer relationships between POI pairs and prototype pairs. Based on the learned bi-level graphs, our model then employs a multi-relational graph network that considers both POI- and prototype-level neighbors, resulting in improved POI representations. Our bi-level structure learning scheme is more robust to data noise and incompleteness, and improves the exploration ability for recommendation by alleviating sparsity issues. Experimental results on three real-world datasets demonstrate the superiority of our model over existing state-of-the-art methods, with a significant improvement in recommendation accuracy and exploration performance.
* Accepted by IEEE Transactions on Knowledge and Data Engineering
MIRAI: Evaluating LLM Agents for Event Forecasting
Jul 01, 2024



Recent advancements in Large Language Models (LLMs) have empowered LLM agents to autonomously collect world information, over which to conduct reasoning to solve complex problems. Given this capability, increasing interests have been put into employing LLM agents for predicting international events, which can influence decision-making and shape policy development on an international scale. Despite such a growing interest, there is a lack of a rigorous benchmark of LLM agents' forecasting capability and reliability. To address this gap, we introduce MIRAI, a novel benchmark designed to systematically evaluate LLM agents as temporal forecasters in the context of international events. Our benchmark features an agentic environment with tools for accessing an extensive database of historical, structured events and textual news articles. We refine the GDELT event database with careful cleaning and parsing to curate a series of relational prediction tasks with varying forecasting horizons, assessing LLM agents' abilities from short-term to long-term forecasting. We further implement APIs to enable LLM agents to utilize different tools via a code-based interface. In summary, MIRAI comprehensively evaluates the agents' capabilities in three dimensions: 1) autonomously source and integrate critical information from large global databases; 2) write codes using domain-specific APIs and libraries for tool-use; and 3) jointly reason over historical knowledge from diverse formats and time to accurately predict future events. Through comprehensive benchmarking, we aim to establish a reliable framework for assessing the capabilities of LLM agents in forecasting international events, thereby contributing to the development of more accurate and trustworthy models for international relation analysis.
Efficient Evolutionary Search Over Chemical Space with Large Language Models
Jun 23, 2024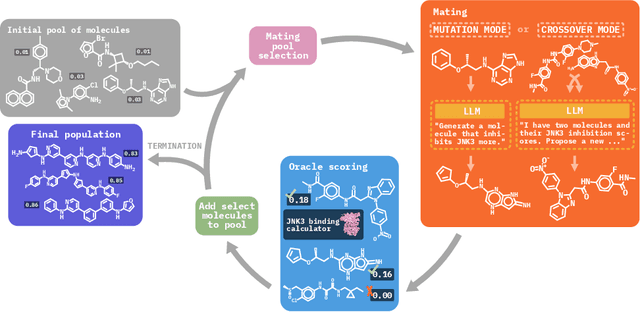
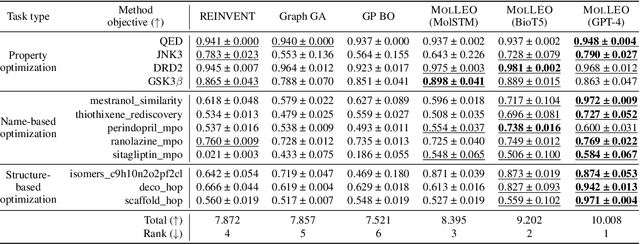
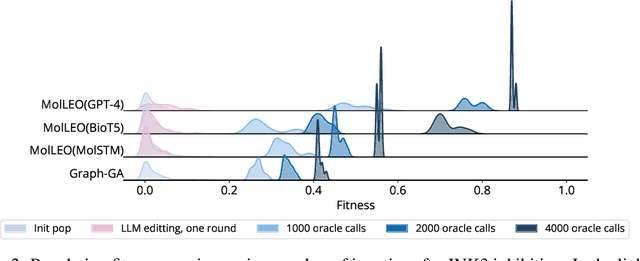
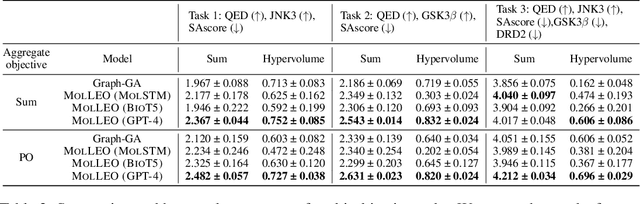
Molecular discovery, when formulated as an optimization problem, presents significant computational challenges because optimization objectives can be non-differentiable. Evolutionary Algorithms (EAs), often used to optimize black-box objectives in molecular discovery, traverse chemical space by performing random mutations and crossovers, leading to a large number of expensive objective evaluations. In this work, we ameliorate this shortcoming by incorporating chemistry-aware Large Language Models (LLMs) into EAs. Namely, we redesign crossover and mutation operations in EAs using LLMs trained on large corpora of chemical information. We perform extensive empirical studies on both commercial and open-source models on multiple tasks involving property optimization, molecular rediscovery, and structure-based drug design, demonstrating that the joint usage of LLMs with EAs yields superior performance over all baseline models across single- and multi-objective settings. We demonstrate that our algorithm improves both the quality of the final solution and convergence speed, thereby reducing the number of required objective evaluations. Our code is available at http://github.com/zoom-wang112358/MOLLEO
BMRetriever: Tuning Large Language Models as Better Biomedical Text Retrievers
Apr 29, 2024



Developing effective biomedical retrieval models is important for excelling at knowledge-intensive biomedical tasks but still challenging due to the deficiency of sufficient publicly annotated biomedical data and computational resources. We present BMRetriever, a series of dense retrievers for enhancing biomedical retrieval via unsupervised pre-training on large biomedical corpora, followed by instruction fine-tuning on a combination of labeled datasets and synthetic pairs. Experiments on 5 biomedical tasks across 11 datasets verify BMRetriever's efficacy on various biomedical applications. BMRetriever also exhibits strong parameter efficiency, with the 410M variant outperforming baselines up to 11.7 times larger, and the 2B variant matching the performance of models with over 5B parameters. The training data and model checkpoints are released at \url{https://huggingface.co/BMRetriever} to ensure transparency, reproducibility, and application to new domains.
An Evaluation of Large Language Models in Bioinformatics Research
Feb 21, 2024



Large language models (LLMs) such as ChatGPT have gained considerable interest across diverse research communities. Their notable ability for text completion and generation has inaugurated a novel paradigm for language-interfaced problem solving. However, the potential and efficacy of these models in bioinformatics remain incompletely explored. In this work, we study the performance LLMs on a wide spectrum of crucial bioinformatics tasks. These tasks include the identification of potential coding regions, extraction of named entities for genes and proteins, detection of antimicrobial and anti-cancer peptides, molecular optimization, and resolution of educational bioinformatics problems. Our findings indicate that, given appropriate prompts, LLMs like GPT variants can successfully handle most of these tasks. In addition, we provide a thorough analysis of their limitations in the context of complicated bioinformatics tasks. In conclusion, we believe that this work can provide new perspectives and motivate future research in the field of LLMs applications, AI for Science and bioinformatics.
GSLB: The Graph Structure Learning Benchmark
Oct 08, 2023Graph Structure Learning (GSL) has recently garnered considerable attention due to its ability to optimize both the parameters of Graph Neural Networks (GNNs) and the computation graph structure simultaneously. Despite the proliferation of GSL methods developed in recent years, there is no standard experimental setting or fair comparison for performance evaluation, which creates a great obstacle to understanding the progress in this field. To fill this gap, we systematically analyze the performance of GSL in different scenarios and develop a comprehensive Graph Structure Learning Benchmark (GSLB) curated from 20 diverse graph datasets and 16 distinct GSL algorithms. Specifically, GSLB systematically investigates the characteristics of GSL in terms of three dimensions: effectiveness, robustness, and complexity. We comprehensively evaluate state-of-the-art GSL algorithms in node- and graph-level tasks, and analyze their performance in robust learning and model complexity. Further, to facilitate reproducible research, we have developed an easy-to-use library for training, evaluating, and visualizing different GSL methods. Empirical results of our extensive experiments demonstrate the ability of GSL and reveal its potential benefits on various downstream tasks, offering insights and opportunities for future research. The code of GSLB is available at: https://github.com/GSL-Benchmark/GSLB.