 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCLORE: Content-Level Optimization for Reasoning Efficiency
May 21, 2026Reinforcement learning post-training has improved the reasoning ability of large language models, but often produces unnecessarily long, repetitive, or semantically opaque reasoning traces. Existing efficient reasoning methods mainly regulate response length through explicit budgets or length-aware rewards, leaving intermediate reasoning content weakly supervised. We propose CLORE, a content-level optimization framework that improves reasoning efficiency by editing correct on-policy rollouts. CLORE uses an external augmentation model to delete repetitive segments, illegible or task-irrelevant content, and superfluous reasoning after the solution is established, while preserving the final answer. The resulting augmented--original pairs are optimized with an auxiliary reference-free DPO objective alongside standard policy-gradient training. By restricting augmentation to correct trajectories and performing local deletion, CLORE keeps edited rollouts close to the policy distribution and mitigates off-policy mismatch. Experiments on DeepSeek-R1-Distill-Qwen-7B and Qwen2.5-Math-7B across five mathematical reasoning benchmarks show that CLORE improves the accuracy--efficiency trade-off and remains compatible with GRPO, DAPO, Training Efficient, and ThinkPrune. Content-level analyses further show that CLORE reduces repetitive reasoning, illegible content, and post-answer exploration, supporting content-level supervision as a complementary direction to length-level control.
Applications of Modular Co-Design for De Novo 3D Molecule Generation
May 23, 2025De novo 3D molecule generation is a pivotal task in drug discovery. However, many recent geometric generative models struggle to produce high-quality 3D structures, even if they maintain 2D validity and topological stability. To tackle this issue and enhance the learning of effective molecular generation dynamics, we present Megalodon-a family of scalable transformer models. These models are enhanced with basic equivariant layers and trained using a joint continuous and discrete denoising co-design objective. We assess Megalodon's performance on established molecule generation benchmarks and introduce new 3D structure benchmarks that evaluate a model's capability to generate realistic molecular structures, particularly focusing on energetics. We show that Megalodon achieves state-of-the-art results in 3D molecule generation, conditional structure generation, and structure energy benchmarks using diffusion and flow matching. Furthermore, doubling the number of parameters in Megalodon to 40M significantly enhances its performance, generating up to 49x more valid large molecules and achieving energy levels that are 2-10x lower than those of the best prior generative models.
GEOM-Drugs Revisited: Toward More Chemically Accurate Benchmarks for 3D Molecule Generation
Apr 30, 2025



Deep generative models have shown significant promise in generating valid 3D molecular structures, with the GEOM-Drugs dataset serving as a key benchmark. However, current evaluation protocols suffer from critical flaws, including incorrect valency definitions, bugs in bond order calculations, and reliance on force fields inconsistent with the reference data. In this work, we revisit GEOM-Drugs and propose a corrected evaluation framework: we identify and fix issues in data preprocessing, construct chemically accurate valency tables, and introduce a GFN2-xTB-based geometry and energy benchmark. We retrain and re-evaluate several leading models under this framework, providing updated performance metrics and practical recommendations for future benchmarking. Our results underscore the need for chemically rigorous evaluation practices in 3D molecular generation. Our recommended evaluation methods and GEOM-Drugs processing scripts are available at https://github.com/isayevlab/geom-drugs-3dgen-evaluation.
A practical guide to machine learning interatomic potentials -- Status and future
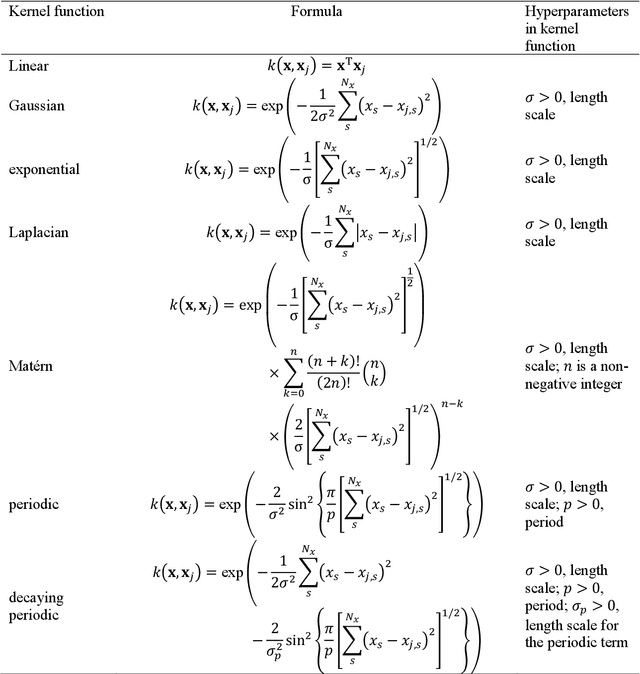
Mar 12, 2025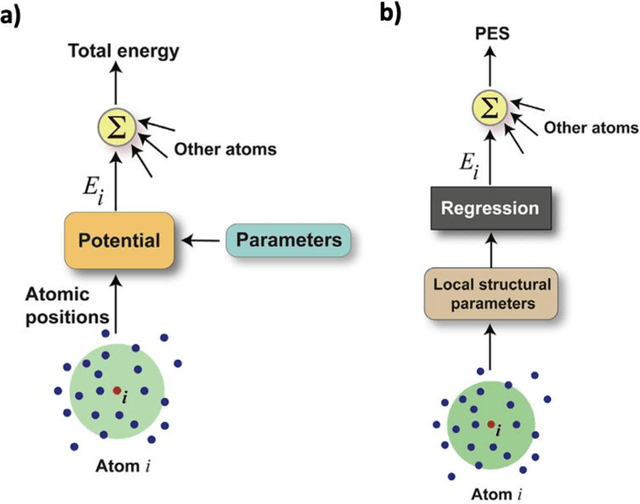

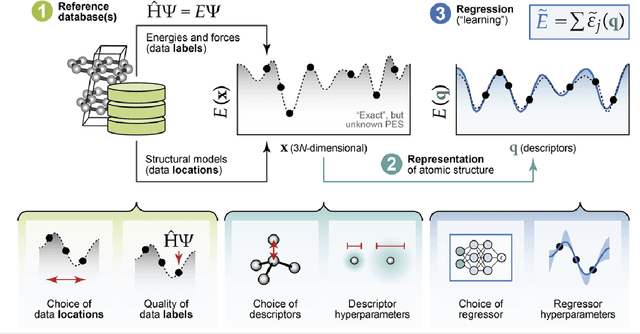
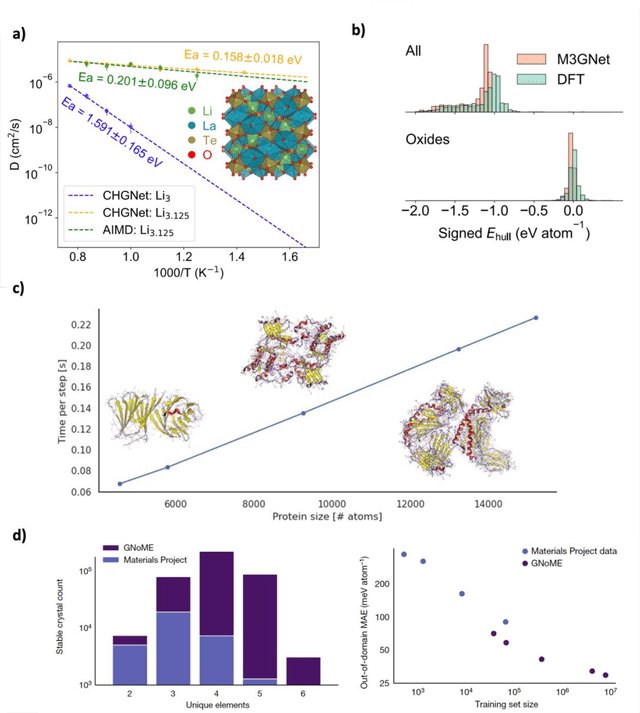
The rapid development and large body of literature on machine learning interatomic potentials (MLIPs) can make it difficult to know how to proceed for researchers who are not experts but wish to use these tools. The spirit of this review is to help such researchers by serving as a practical, accessible guide to the state-of-the-art in MLIPs. This review paper covers a broad range of topics related to MLIPs, including (i) central aspects of how and why MLIPs are enablers of many exciting advancements in molecular modeling, (ii) the main underpinnings of different types of MLIPs, including their basic structure and formalism, (iii) the potentially transformative impact of universal MLIPs for both organic and inorganic systems, including an overview of the most recent advances, capabilities, downsides, and potential applications of this nascent class of MLIPs, (iv) a practical guide for estimating and understanding the execution speed of MLIPs, including guidance for users based on hardware availability, type of MLIP used, and prospective simulation size and time, (v) a manual for what MLIP a user should choose for a given application by considering hardware resources, speed requirements, energy and force accuracy requirements, as well as guidance for choosing pre-trained potentials or fitting a new potential from scratch, (vi) discussion around MLIP infrastructure, including sources of training data, pre-trained potentials, and hardware resources for training, (vii) summary of some key limitations of present MLIPs and current approaches to mitigate such limitations, including methods of including long-range interactions, handling magnetic systems, and treatment of excited states, and finally (viii) we finish with some more speculative thoughts on what the future holds for the development and application of MLIPs over the next 3-10+ years.
MLatom 3: Platform for machine learning-enhanced computational chemistry simulations and workflows
Oct 31, 2023
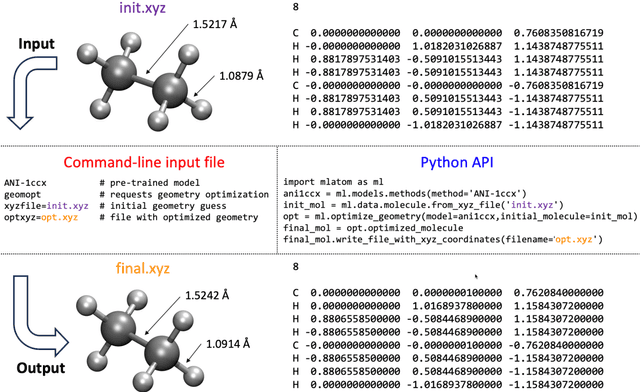
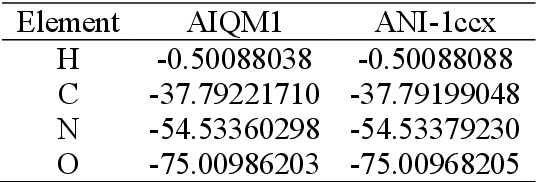
Machine learning (ML) is increasingly becoming a common tool in computational chemistry. At the same time, the rapid development of ML methods requires a flexible software framework for designing custom workflows. MLatom 3 is a program package designed to leverage the power of ML to enhance typical computational chemistry simulations and to create complex workflows. This open-source package provides plenty of choice to the users who can run simulations with the command line options, input files, or with scripts using MLatom as a Python package, both on their computers and on the online XACS cloud computing at XACScloud.com. Computational chemists can calculate energies and thermochemical properties, optimize geometries, run molecular and quantum dynamics, and simulate (ro)vibrational, one-photon UV/vis absorption, and two-photon absorption spectra with ML, quantum mechanical, and combined models. The users can choose from an extensive library of methods containing pre-trained ML models and quantum mechanical approximations such as AIQM1 approaching coupled-cluster accuracy. The developers can build their own models using various ML algorithms. The great flexibility of MLatom is largely due to the extensive use of the interfaces to many state-of-the-art software packages and libraries.
Learning Over Molecular Conformer Ensembles: Datasets and Benchmarks
Sep 29, 2023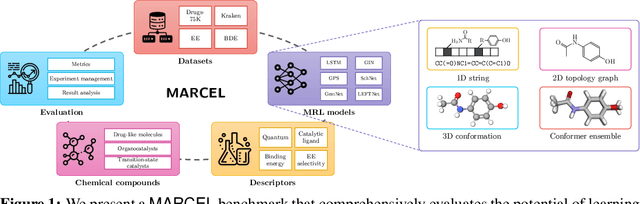
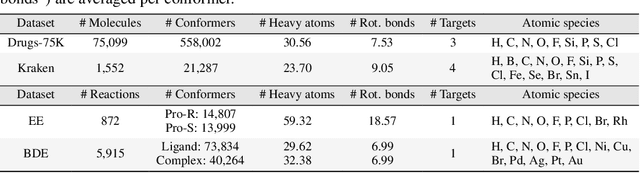
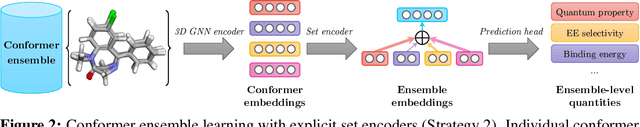
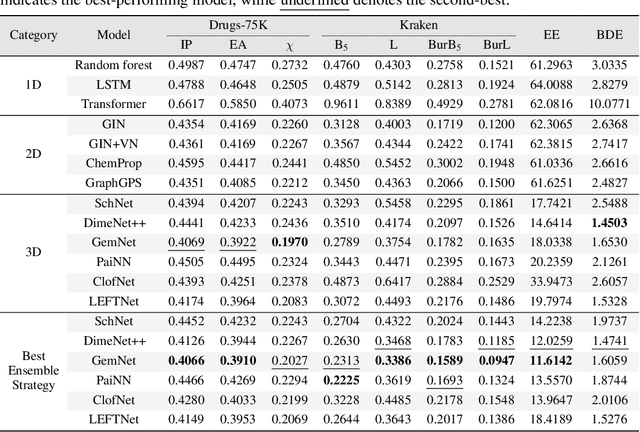
Molecular Representation Learning (MRL) has proven impactful in numerous biochemical applications such as drug discovery and enzyme design. While Graph Neural Networks (GNNs) are effective at learning molecular representations from a 2D molecular graph or a single 3D structure, existing works often overlook the flexible nature of molecules, which continuously interconvert across conformations via chemical bond rotations and minor vibrational perturbations. To better account for molecular flexibility, some recent works formulate MRL as an ensemble learning problem, focusing on explicitly learning from a set of conformer structures. However, most of these studies have limited datasets, tasks, and models. In this work, we introduce the first MoleculAR Conformer Ensemble Learning (MARCEL) benchmark to thoroughly evaluate the potential of learning on conformer ensembles and suggest promising research directions. MARCEL includes four datasets covering diverse molecule- and reaction-level properties of chemically diverse molecules including organocatalysts and transition-metal catalysts, extending beyond the scope of common GNN benchmarks that are confined to drug-like molecules. In addition, we conduct a comprehensive empirical study, which benchmarks representative 1D, 2D, and 3D molecular representation learning models, along with two strategies that explicitly incorporate conformer ensembles into 3D MRL models. Our findings reveal that direct learning from an accessible conformer space can improve performance on a variety of tasks and models.
Simulation Intelligence: Towards a New Generation of Scientific Methods
Dec 06, 2021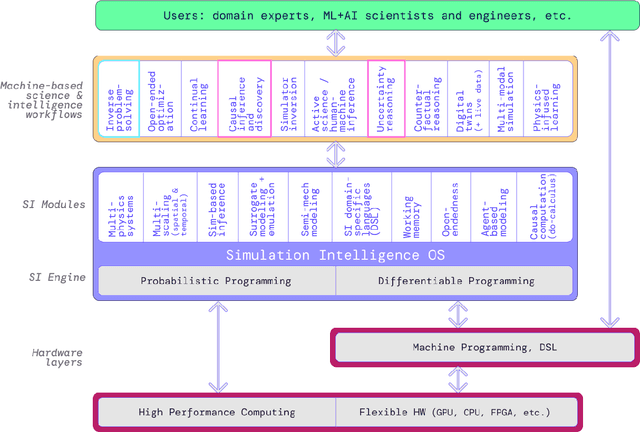
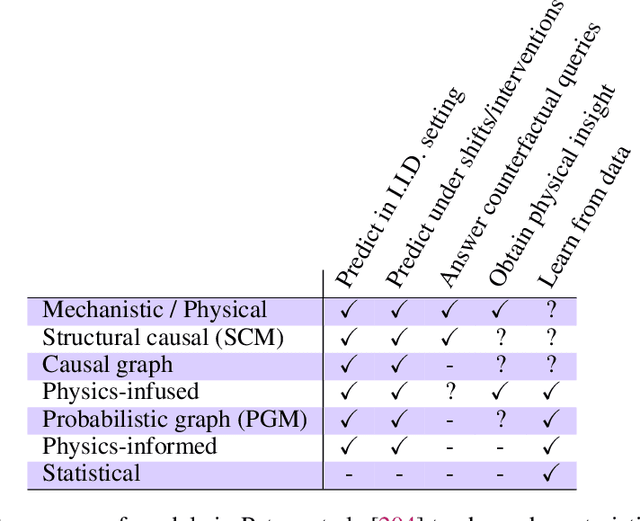
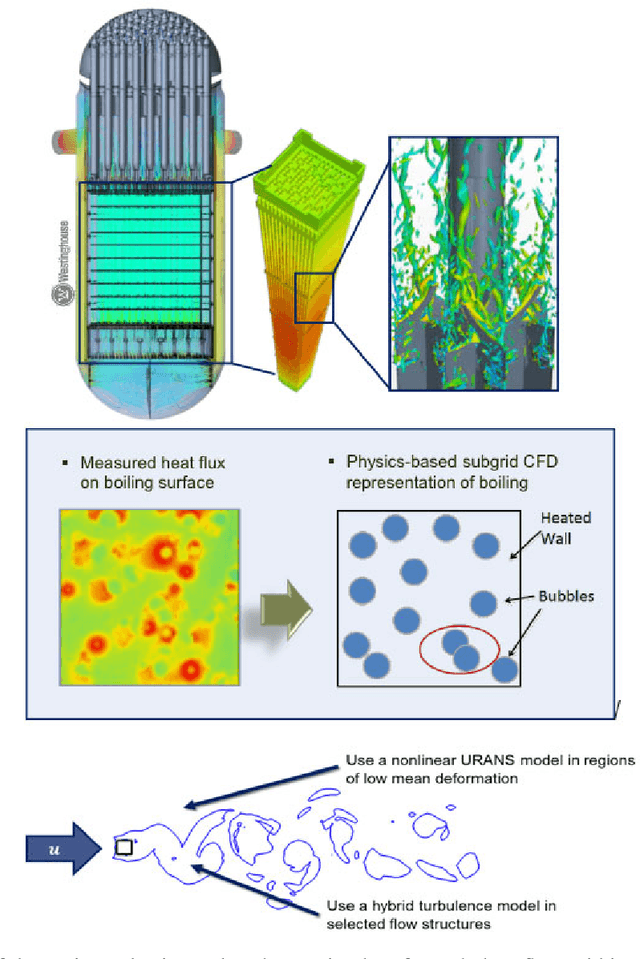
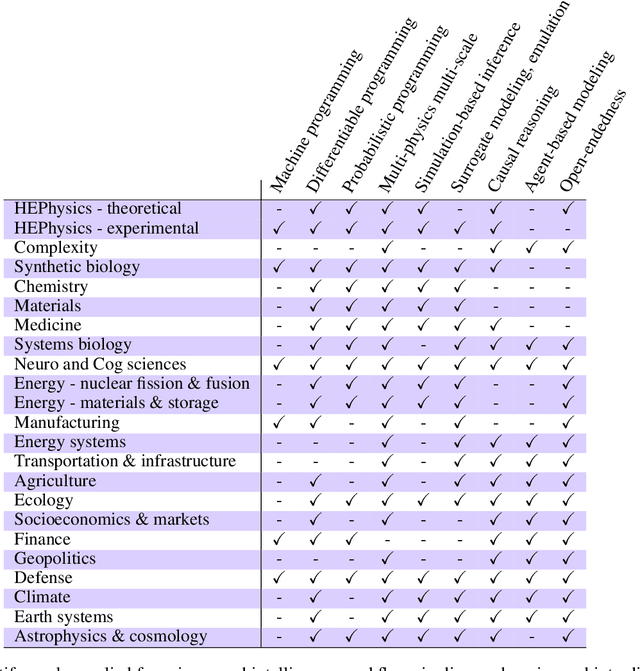
The original "Seven Motifs" set forth a roadmap of essential methods for the field of scientific computing, where a motif is an algorithmic method that captures a pattern of computation and data movement. We present the "Nine Motifs of Simulation Intelligence", a roadmap for the development and integration of the essential algorithms necessary for a merger of scientific computing, scientific simulation, and artificial intelligence. We call this merger simulation intelligence (SI), for short. We argue the motifs of simulation intelligence are interconnected and interdependent, much like the components within the layers of an operating system. Using this metaphor, we explore the nature of each layer of the simulation intelligence operating system stack (SI-stack) and the motifs therein: (1) Multi-physics and multi-scale modeling; (2) Surrogate modeling and emulation; (3) Simulation-based inference; (4) Causal modeling and inference; (5) Agent-based modeling; (6) Probabilistic programming; (7) Differentiable programming; (8) Open-ended optimization; (9) Machine programming. We believe coordinated efforts between motifs offers immense opportunity to accelerate scientific discovery, from solving inverse problems in synthetic biology and climate science, to directing nuclear energy experiments and predicting emergent behavior in socioeconomic settings. We elaborate on each layer of the SI-stack, detailing the state-of-art methods, presenting examples to highlight challenges and opportunities, and advocating for specific ways to advance the motifs and the synergies from their combinations. Advancing and integrating these technologies can enable a robust and efficient hypothesis-simulation-analysis type of scientific method, which we introduce with several use-cases for human-machine teaming and automated science.
Impressive computational acceleration by using machine learning for 2-dimensional super-lubricant materials discovery
Nov 20, 2019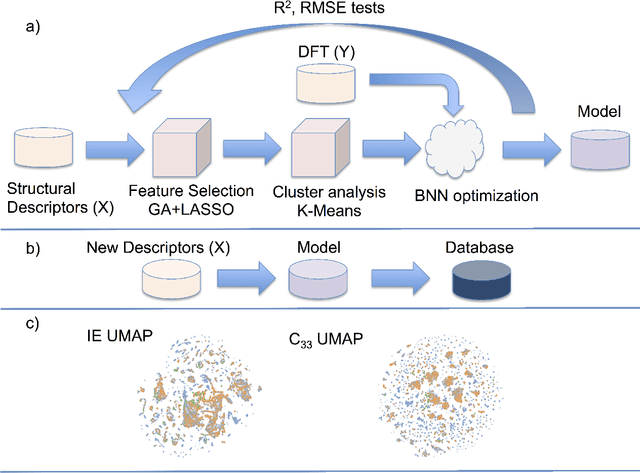
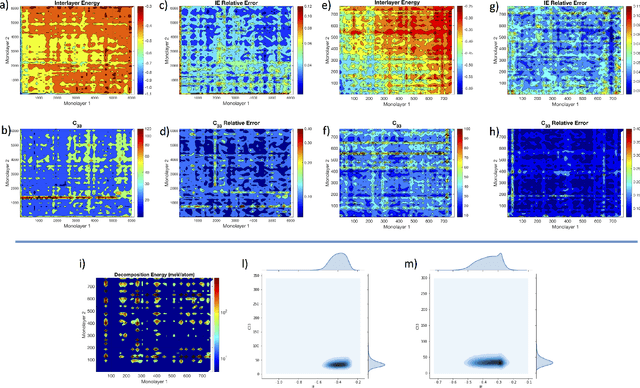

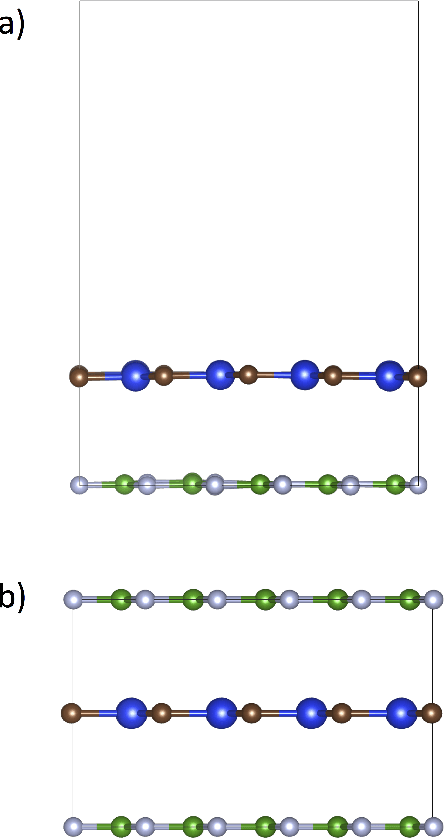
The screening of novel materials is an important topic in the field of materials science. Although traditional computational modeling, especially first-principles approaches, is a very useful and accurate tool to predict the properties of novel materials, it still demands extensive and expensive state-of-the-art computational resources. Additionally, they can be often extremely time consuming. We describe a time and resource-efficient machine learning approach to create a large dataset of structural properties of van der Waals layered structures. In particular, we focus on the interlayer energy and the elastic constant of layered materials composed of two different 2-dimensional (2D) structures, that are important for novel solid lubricant and super-lubricant materials. We show that machine learning models can recapitulate results of computationally expansive approaches (i.e. density functional theory) with high accuracy.
MolecularRNN: Generating realistic molecular graphs with optimized properties
May 31, 2019


Designing new molecules with a set of predefined properties is a core problem in modern drug discovery and development. There is a growing need for de-novo design methods that would address this problem. We present MolecularRNN, the graph recurrent generative model for molecular structures. Our model generates diverse realistic molecular graphs after likelihood pretraining on a big database of molecules. We perform an analysis of our pretrained models on large-scale generated datasets of 1 million samples. Further, the model is tuned with policy gradient algorithm, provided a critic that estimates the reward for the property of interest. We show a significant distribution shift to the desired range for lipophilicity, drug-likeness, and melting point outperforming state-of-the-art works. With the use of rejection sampling based on valency constraints, our model yields 100% validity. Moreover, we show that invalid molecules provide a rich signal to the model through the use of structure penalty in our reinforcement learning pipeline.
Deep Reinforcement Learning for De-Novo Drug Design
May 31, 2018We propose a novel computational strategy for de novo design of molecules with desired properties termed ReLeaSE (Reinforcement Learning for Structural Evolution). Based on deep and reinforcement learning approaches, ReLeaSE integrates two deep neural networks - generative and predictive - that are trained separately but employed jointly to generate novel targeted chemical libraries. ReLeaSE employs simple representation of molecules by their SMILES strings only. Generative models are trained with stack-augmented memory network to produce chemically feasible SMILES strings, and predictive models are derived to forecast the desired properties of the de novo generated compounds. In the first phase of the method, generative and predictive models are trained separately with a supervised learning algorithm. In the second phase, both models are trained jointly with the reinforcement learning approach to bias the generation of new chemical structures towards those with the desired physical and/or biological properties. In the proof-of-concept study, we have employed the ReLeaSE method to design chemical libraries with a bias toward structural complexity or biased toward compounds with either maximal, minimal, or specific range of physical properties such as melting point or hydrophobicity, as well as to develop novel putative inhibitors of JAK2. The approach proposed herein can find a general use for generating targeted chemical libraries of novel compounds optimized for either a single desired property or multiple properties.