 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Descriptor Is All You Need: Accurate Machine Learning of Nonadiabatic Coupling Vectors
May 29, 2025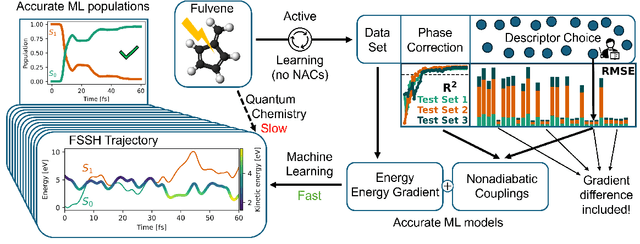

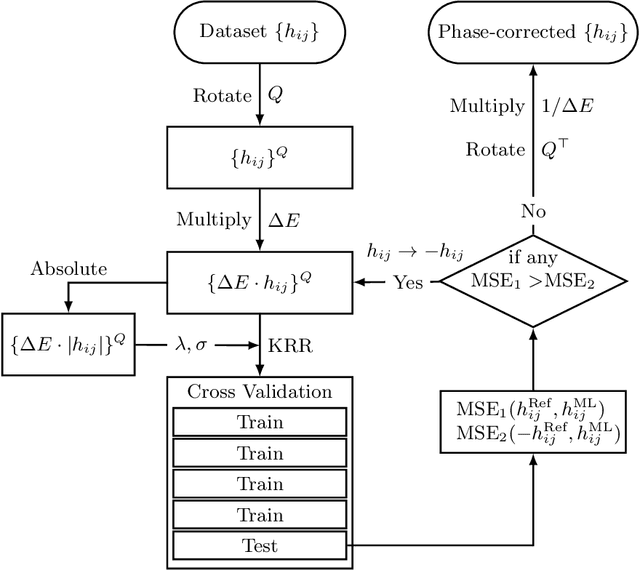
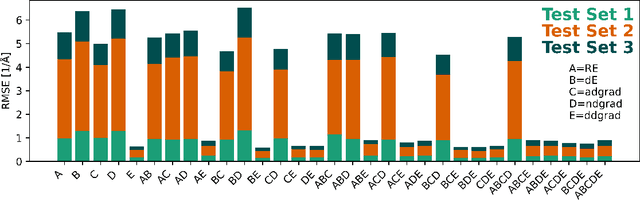
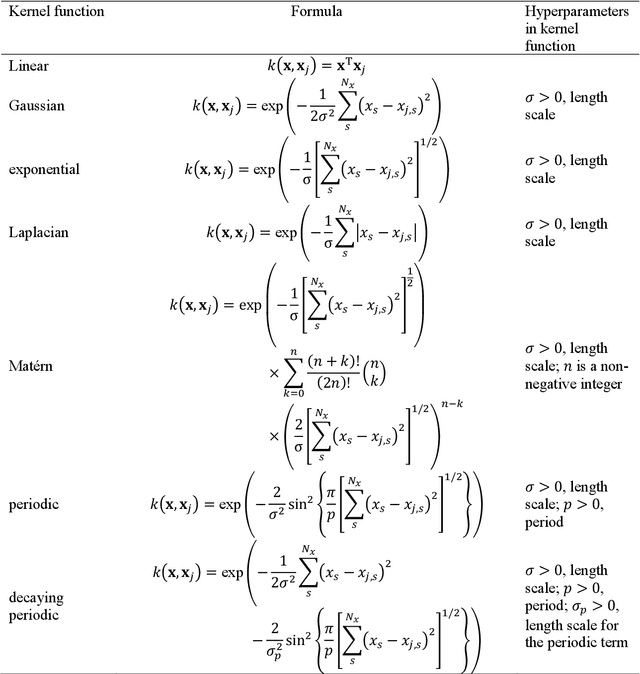
Nonadiabatic couplings (NACs) play a crucial role in modeling photochemical and photophysical processes with methods such as the widely used fewest-switches surface hopping (FSSH). There is therefore a strong incentive to machine learn NACs for accelerating simulations. However, this is challenging due to NACs' vectorial, double-valued character and the singularity near a conical intersection seam. For the first time, we design NAC-specific descriptors based on our domain expertise and show that they allow learning NACs with never-before-reported accuracy of $R^2$ exceeding 0.99. The key to success is also our new ML phase-correction procedure. We demonstrate the efficiency and robustness of our approach on a prototypical example of fully ML-driven FSSH simulations of fulvene targeting the SA-2-CASSCF(6,6) electronic structure level. This ML-FSSH dynamics leads to an accurate description of $S_1$ decay while reducing error bars by allowing the execution of a large ensemble of trajectories. Our implementations are available in open-source MLatom.
Artificial Intelligence for Direct Prediction of Molecular Dynamics Across Chemical Space
May 22, 2025Molecular dynamics (MD) is a powerful tool for exploring the behavior of atomistic systems, but its reliance on sequential numerical integration limits simulation efficiency. We present MDtrajNet-1, a foundational AI model that directly generates MD trajectories across chemical space, bypassing force calculations and integration. This approach accelerates simulations by up to two orders of magnitude compared to traditional MD, even those enhanced by machine-learning interatomic potentials. MDtrajNet-1 combines equivariant neural networks with a Transformer-based architecture to achieve strong accuracy and transferability in predicting long-time trajectories for both known and unseen systems. Remarkably, the errors of the trajectories generated by MDtrajNet-1 for various molecular systems are close to those of the conventional ab initio MD. The model's flexible design supports diverse application scenarios, including different statistical ensembles, boundary conditions, and interaction types. By overcoming the intrinsic speed barrier of conventional MD, MDtrajNet-1 opens new frontiers in efficient and scalable atomistic simulations.
Aitomia: Your Intelligent Assistant for AI-Driven Atomistic and Quantum Chemical Simulations
May 13, 2025We have developed Aitomia - a platform powered by AI to assist in performing AI-driven atomistic and quantum chemical (QC) simulations. This intelligent assistant platform is equipped with chatbots and AI agents to help experts and guide non-experts in setting up and running the atomistic simulations, monitoring their computation status, analyzing the simulation results, and summarizing them for the user in text and graphical forms. We achieve these goals by exploiting fine-tuned open-source large language models (LLMs), rule-based agents, and a retrieval-augmented generation (RAG) system. Aitomia leverages the versatility of our MLatom ecosystem for AI-enhanced computational chemistry. This intelligent assistant is going to be integrated into the Aitomistic Hub and XACS online computing services, with some functionality already publicly available as described at http://mlatom.com/aitomia. Aitomia is expected to lower the barrier to performing atomistic simulations, accelerating research and development in the relevant fields.
All-in-one foundational models learning across quantum chemical levels
Sep 18, 2024Machine learning (ML) potentials typically target a single quantum chemical (QC) level while the ML models developed for multi-fidelity learning have not been shown to provide scalable solutions for foundational models. Here we introduce the all-in-one (AIO) ANI model architecture based on multimodal learning which can learn an arbitrary number of QC levels. Our all-in-one learning approach offers a more general and easier-to-use alternative to transfer learning. We use it to train the AIO-ANI-UIP foundational model with the generalization capability comparable to semi-empirical GFN2-xTB and DFT with a double-zeta basis set for organic molecules. We show that the AIO-ANI model can learn across different QC levels ranging from semi-empirical to density functional theory to coupled cluster. We also use AIO models to design the foundational model {\Delta}-AIO-ANI based on {\Delta}-learning with increased accuracy and robustness compared to AIO-ANI-UIP. The code and the foundational models are available at https://github.com/dralgroup/aio-ani; they will be integrated into the universal and updatable AI-enhanced QM (UAIQM) library and made available in the MLatom package so that they can be used online at the XACS cloud computing platform (see https://github.com/dralgroup/mlatom for updates).
Physics-informed active learning for accelerating quantum chemical simulations
Apr 18, 2024Quantum chemical simulations can be greatly accelerated by constructing machine learning potentials, which is often done using active learning (AL). The usefulness of the constructed potentials is often limited by the high effort required and their insufficient robustness in the simulations. Here we introduce the end-to-end AL for constructing robust data-efficient potentials with affordable investment of time and resources and minimum human interference. Our AL protocol is based on the physics-informed sampling of training points, automatic selection of initial data, and uncertainty quantification. The versatility of this protocol is shown in our implementation of quasi-classical molecular dynamics for simulating vibrational spectra, conformer search of a key biochemical molecule, and time-resolved mechanism of the Diels-Alder reaction. These investigations took us days instead of weeks of pure quantum chemical calculations on a high-performance computing cluster.
MLatom 3: Platform for machine learning-enhanced computational chemistry simulations and workflows
Oct 31, 2023
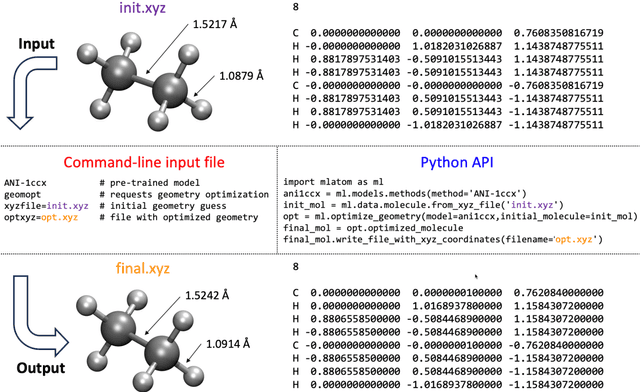
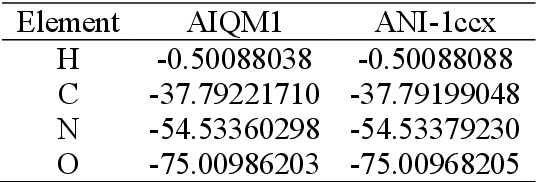
Machine learning (ML) is increasingly becoming a common tool in computational chemistry. At the same time, the rapid development of ML methods requires a flexible software framework for designing custom workflows. MLatom 3 is a program package designed to leverage the power of ML to enhance typical computational chemistry simulations and to create complex workflows. This open-source package provides plenty of choice to the users who can run simulations with the command line options, input files, or with scripts using MLatom as a Python package, both on their computers and on the online XACS cloud computing at XACScloud.com. Computational chemists can calculate energies and thermochemical properties, optimize geometries, run molecular and quantum dynamics, and simulate (ro)vibrational, one-photon UV/vis absorption, and two-photon absorption spectra with ML, quantum mechanical, and combined models. The users can choose from an extensive library of methods containing pre-trained ML models and quantum mechanical approximations such as AIQM1 approaching coupled-cluster accuracy. The developers can build their own models using various ML algorithms. The great flexibility of MLatom is largely due to the extensive use of the interfaces to many state-of-the-art software packages and libraries.