 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeKANEL: Kolmogorov-Arnold Network Ensemble Learning Enables Early Hit Enrichment in High-Throughput Virtual Screening
Mar 25, 2026Machine learning models of chemical bioactivity are increasingly used for prioritizing a small number of compounds in virtual screening libraries for experimental follow-up. In these applications, assessing model accuracy by early hit enrichment such as Positive Predicted Value (PPV) calculated for top N hits (PPV@N) is more appropriate and actionable than traditional global metrics such as AUC. We present KANEL, an ensemble workflow that combines interpretable Kolmogorov-Arnold Networks (KANs) with XGBoost, random forest, and multilayer perceptron models trained on complementary molecular representations (LillyMol descriptors, RDKit-derived descriptors, and Morgan fingerprints).
Explainable Enrichment-Driven GrAph Reasoner (EDGAR) for Large Knowledge Graphs with Applications in Drug Repurposing
Sep 27, 2024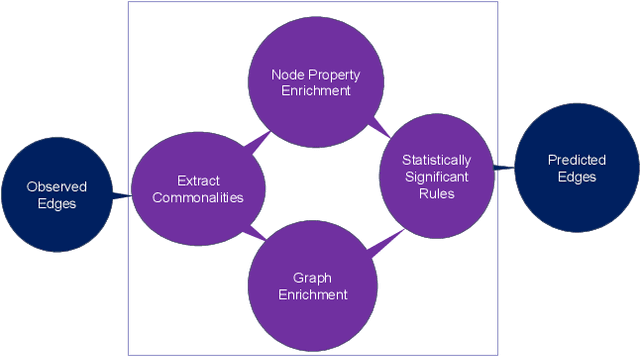
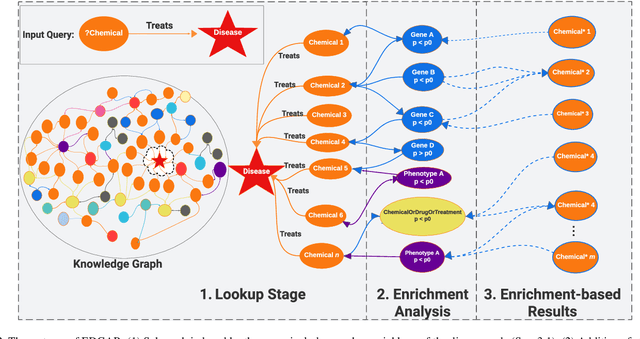

Knowledge graphs (KGs) represent connections and relationships between real-world entities. We propose a link prediction framework for KGs named Enrichment-Driven GrAph Reasoner (EDGAR), which infers new edges by mining entity-local rules. This approach leverages enrichment analysis, a well-established statistical method used to identify mechanisms common to sets of differentially expressed genes. EDGAR's inference results are inherently explainable and rankable, with p-values indicating the statistical significance of each enrichment-based rule. We demonstrate the framework's effectiveness on a large-scale biomedical KG, ROBOKOP, focusing on drug repurposing for Alzheimer disease (AD) as a case study. Initially, we extracted 14 known drugs from the KG and identified 20 contextual biomarkers through enrichment analysis, revealing functional pathways relevant to shared drug efficacy for AD. Subsequently, using the top 1000 enrichment results, our system identified 1246 additional drug candidates for AD treatment. The top 10 candidates were validated using evidence from medical literature. EDGAR is deployed within ROBOKOP, complete with a web user interface. This is the first study to apply enrichment analysis to large graph completion and drug repurposing.
EMOE: Expansive Matching of Experts for Robust Uncertainty Based Rejection
Jun 03, 2024



Expansive Matching of Experts (EMOE) is a novel method that utilizes support-expanding, extrapolatory pseudo-labeling to improve prediction and uncertainty based rejection on out-of-distribution (OOD) points. We propose an expansive data augmentation technique that generates OOD instances in a latent space, and an empirical trial based approach to filter out augmented expansive points for pseudo-labeling. EMOE utilizes a diverse set of multiple base experts as pseudo-labelers on the augmented data to improve OOD performance through a shared MLP with multiple heads (one per expert). We demonstrate that EMOE achieves superior performance compared to state-of-the-art methods on tabular data.
Utilizing Low-Dimensional Molecular Embeddings for Rapid Chemical Similarity Search
Feb 12, 2024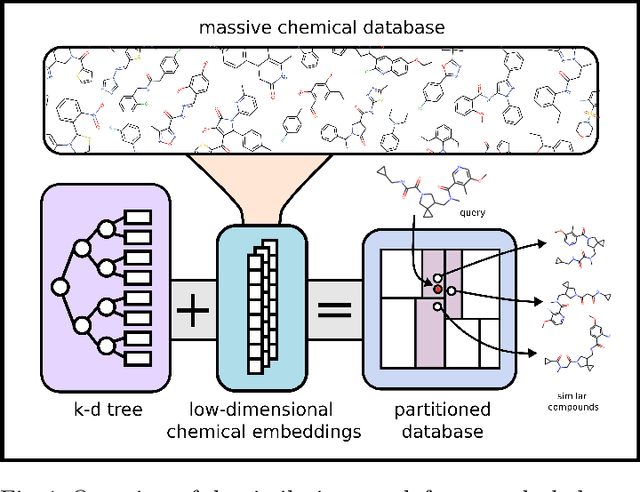
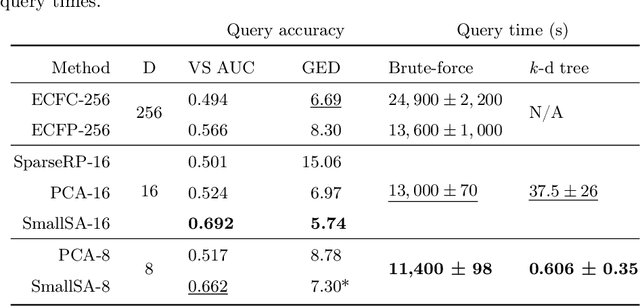
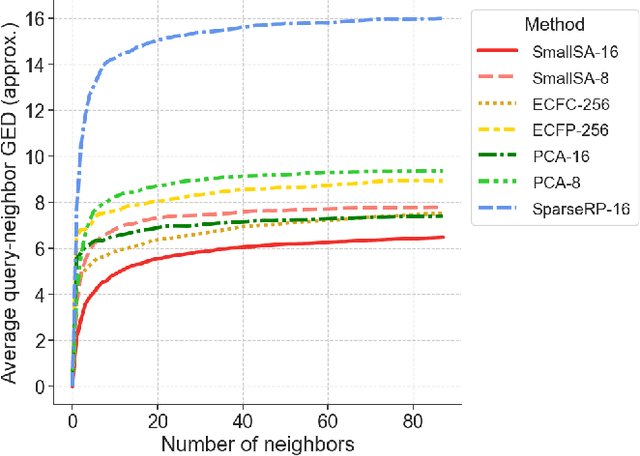
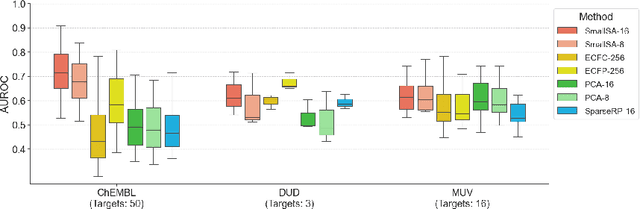
Nearest neighbor-based similarity searching is a common task in chemistry, with notable use cases in drug discovery. Yet, some of the most commonly used approaches for this task still leverage a brute-force approach. In practice this can be computationally costly and overly time-consuming, due in part to the sheer size of modern chemical databases. Previous computational advancements for this task have generally relied on improvements to hardware or dataset-specific tricks that lack generalizability. Approaches that leverage lower-complexity searching algorithms remain relatively underexplored. However, many of these algorithms are approximate solutions and/or struggle with typical high-dimensional chemical embeddings. Here we evaluate whether a combination of low-dimensional chemical embeddings and a k-d tree data structure can achieve fast nearest neighbor queries while maintaining performance on standard chemical similarity search benchmarks. We examine different dimensionality reductions of standard chemical embeddings as well as a learned, structurally-aware embedding -- SmallSA -- for this task. With this framework, searches on over one billion chemicals execute in less than a second on a single CPU core, five orders of magnitude faster than the brute-force approach. We also demonstrate that SmallSA achieves competitive performance on chemical similarity benchmarks.
SALSA: Semantically-Aware Latent Space Autoencoder
Oct 04, 2023In deep learning for drug discovery, chemical data are often represented as simplified molecular-input line-entry system (SMILES) sequences which allow for straightforward implementation of natural language processing methodologies, one being the sequence-to-sequence autoencoder. However, we observe that training an autoencoder solely on SMILES is insufficient to learn molecular representations that are semantically meaningful, where semantics are defined by the structural (graph-to-graph) similarities between molecules. We demonstrate by example that autoencoders may map structurally similar molecules to distant codes, resulting in an incoherent latent space that does not respect the structural similarities between molecules. To address this shortcoming we propose Semantically-Aware Latent Space Autoencoder (SALSA), a transformer-autoencoder modified with a contrastive task, tailored specifically to learn graph-to-graph similarity between molecules. Formally, the contrastive objective is to map structurally similar molecules (separated by a single graph edit) to nearby codes in the latent space. To accomplish this, we generate a novel dataset comprised of sets of structurally similar molecules and opt for a supervised contrastive loss that is able to incorporate full sets of positive samples. We compare SALSA to its ablated counterparts, and show empirically that the composed training objective (reconstruction and contrastive task) leads to a higher quality latent space that is more 1) structurally-aware, 2) semantically continuous, and 3) property-aware.
Deep Reinforcement Learning for De-Novo Drug Design
May 31, 2018We propose a novel computational strategy for de novo design of molecules with desired properties termed ReLeaSE (Reinforcement Learning for Structural Evolution). Based on deep and reinforcement learning approaches, ReLeaSE integrates two deep neural networks - generative and predictive - that are trained separately but employed jointly to generate novel targeted chemical libraries. ReLeaSE employs simple representation of molecules by their SMILES strings only. Generative models are trained with stack-augmented memory network to produce chemically feasible SMILES strings, and predictive models are derived to forecast the desired properties of the de novo generated compounds. In the first phase of the method, generative and predictive models are trained separately with a supervised learning algorithm. In the second phase, both models are trained jointly with the reinforcement learning approach to bias the generation of new chemical structures towards those with the desired physical and/or biological properties. In the proof-of-concept study, we have employed the ReLeaSE method to design chemical libraries with a bias toward structural complexity or biased toward compounds with either maximal, minimal, or specific range of physical properties such as melting point or hydrophobicity, as well as to develop novel putative inhibitors of JAK2. The approach proposed herein can find a general use for generating targeted chemical libraries of novel compounds optimized for either a single desired property or multiple properties.