 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCorrection of Automatic Speech Recognition with Transformer Sequence-to-sequence Model
Oct 23, 2019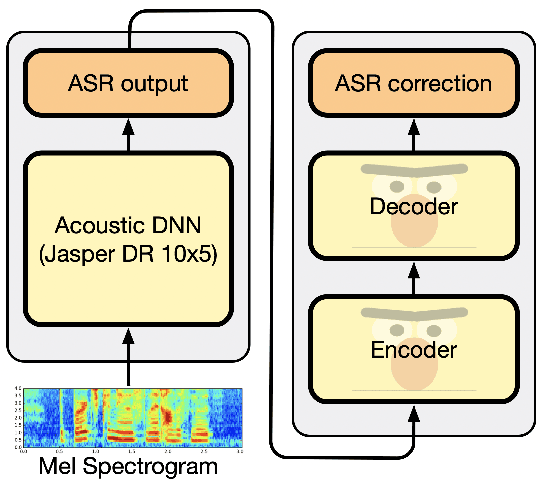
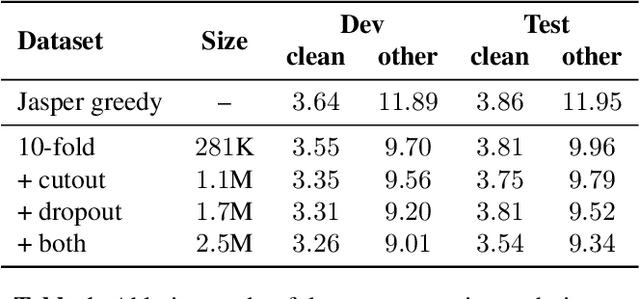
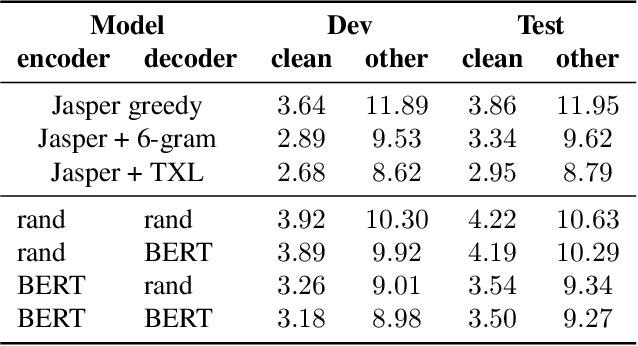
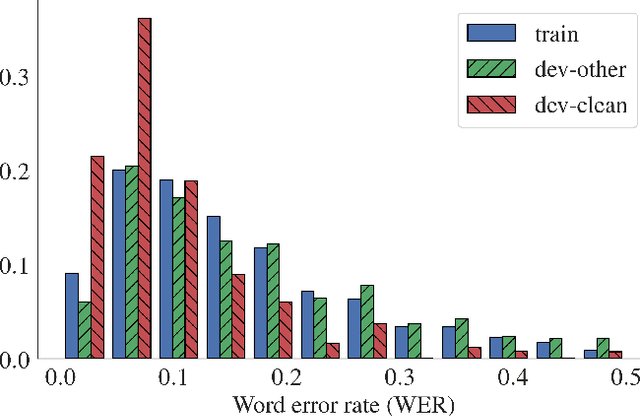
In this work, we introduce a simple yet efficient post-processing model for automatic speech recognition (ASR). Our model has Transformer-based encoder-decoder architecture which "translates" ASR model output into grammatically and semantically correct text. We investigate different strategies for regularizing and optimizing the model and show that extensive data augmentation and the initialization with pre-trained weights are required to achieve good performance. On the LibriSpeech benchmark, our method demonstrates significant improvement in word error rate over the baseline acoustic model with greedy decoding, especially on much noisier dev-other and test-other portions of the evaluation dataset. Our model also outperforms baseline with 6-gram language model re-scoring and approaches the performance of re-scoring with Transformer-XL neural language model.
NeMo: a toolkit for building AI applications using Neural Modules
Sep 14, 2019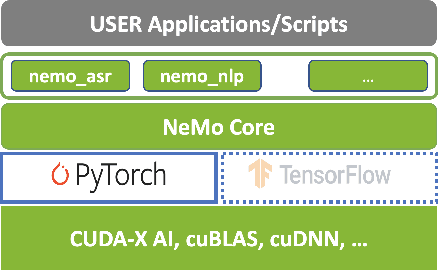

NeMo (Neural Modules) is a Python framework-agnostic toolkit for creating AI applications through re-usability, abstraction, and composition. NeMo is built around neural modules, conceptual blocks of neural networks that take typed inputs and produce typed outputs. Such modules typically represent data layers, encoders, decoders, language models, loss functions, or methods of combining activations. NeMo makes it easy to combine and re-use these building blocks while providing a level of semantic correctness checking via its neural type system. The toolkit comes with extendable collections of pre-built modules for automatic speech recognition and natural language processing. Furthermore, NeMo provides built-in support for distributed training and mixed precision on latest NVIDIA GPUs. NeMo is open-source https://github.com/NVIDIA/NeMo
MolecularRNN: Generating realistic molecular graphs with optimized properties
May 31, 2019
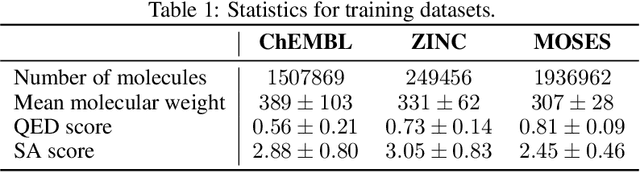


Designing new molecules with a set of predefined properties is a core problem in modern drug discovery and development. There is a growing need for de-novo design methods that would address this problem. We present MolecularRNN, the graph recurrent generative model for molecular structures. Our model generates diverse realistic molecular graphs after likelihood pretraining on a big database of molecules. We perform an analysis of our pretrained models on large-scale generated datasets of 1 million samples. Further, the model is tuned with policy gradient algorithm, provided a critic that estimates the reward for the property of interest. We show a significant distribution shift to the desired range for lipophilicity, drug-likeness, and melting point outperforming state-of-the-art works. With the use of rejection sampling based on valency constraints, our model yields 100% validity. Moreover, we show that invalid molecules provide a rich signal to the model through the use of structure penalty in our reinforcement learning pipeline.
Deep Reinforcement Learning for De-Novo Drug Design
May 31, 2018We propose a novel computational strategy for de novo design of molecules with desired properties termed ReLeaSE (Reinforcement Learning for Structural Evolution). Based on deep and reinforcement learning approaches, ReLeaSE integrates two deep neural networks - generative and predictive - that are trained separately but employed jointly to generate novel targeted chemical libraries. ReLeaSE employs simple representation of molecules by their SMILES strings only. Generative models are trained with stack-augmented memory network to produce chemically feasible SMILES strings, and predictive models are derived to forecast the desired properties of the de novo generated compounds. In the first phase of the method, generative and predictive models are trained separately with a supervised learning algorithm. In the second phase, both models are trained jointly with the reinforcement learning approach to bias the generation of new chemical structures towards those with the desired physical and/or biological properties. In the proof-of-concept study, we have employed the ReLeaSE method to design chemical libraries with a bias toward structural complexity or biased toward compounds with either maximal, minimal, or specific range of physical properties such as melting point or hydrophobicity, as well as to develop novel putative inhibitors of JAK2. The approach proposed herein can find a general use for generating targeted chemical libraries of novel compounds optimized for either a single desired property or multiple properties.