 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMolecularRNN: Generating realistic molecular graphs with optimized properties
Paper and Code

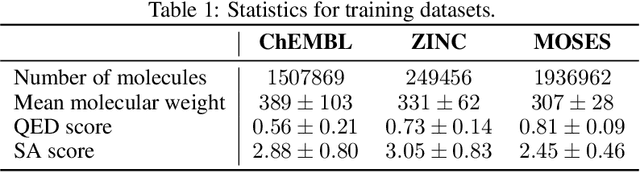


Designing new molecules with a set of predefined properties is a core problem in modern drug discovery and development. There is a growing need for de-novo design methods that would address this problem. We present MolecularRNN, the graph recurrent generative model for molecular structures. Our model generates diverse realistic molecular graphs after likelihood pretraining on a big database of molecules. We perform an analysis of our pretrained models on large-scale generated datasets of 1 million samples. Further, the model is tuned with policy gradient algorithm, provided a critic that estimates the reward for the property of interest. We show a significant distribution shift to the desired range for lipophilicity, drug-likeness, and melting point outperforming state-of-the-art works. With the use of rejection sampling based on valency constraints, our model yields 100% validity. Moreover, we show that invalid molecules provide a rich signal to the model through the use of structure penalty in our reinforcement learning pipeline.