 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeUtilizing Low-Dimensional Molecular Embeddings for Rapid Chemical Similarity Search
Feb 12, 2024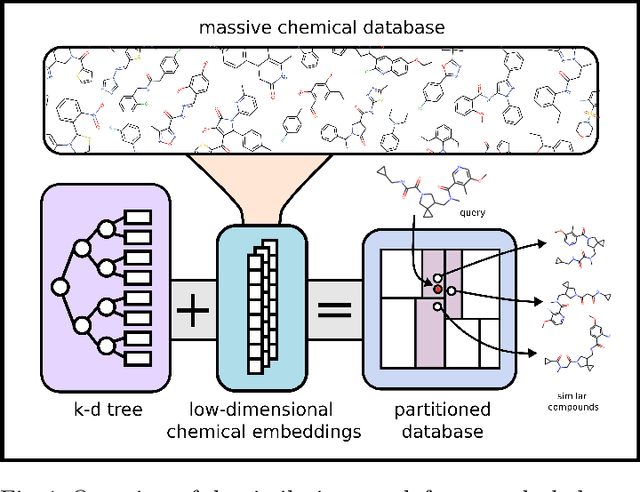
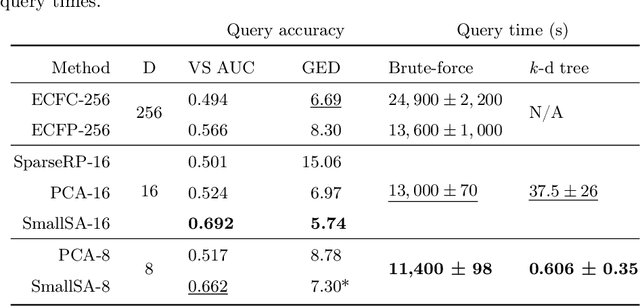
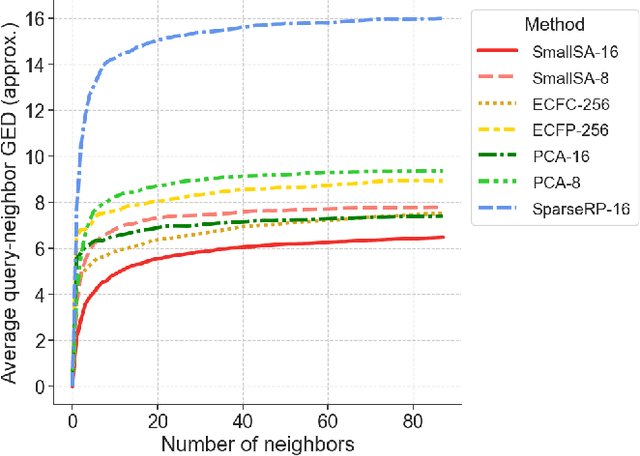
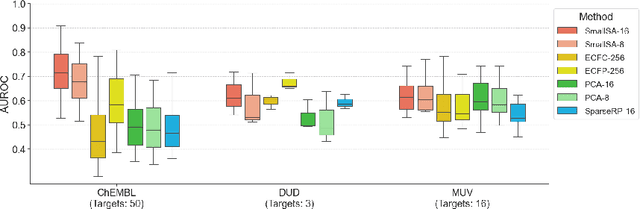
Nearest neighbor-based similarity searching is a common task in chemistry, with notable use cases in drug discovery. Yet, some of the most commonly used approaches for this task still leverage a brute-force approach. In practice this can be computationally costly and overly time-consuming, due in part to the sheer size of modern chemical databases. Previous computational advancements for this task have generally relied on improvements to hardware or dataset-specific tricks that lack generalizability. Approaches that leverage lower-complexity searching algorithms remain relatively underexplored. However, many of these algorithms are approximate solutions and/or struggle with typical high-dimensional chemical embeddings. Here we evaluate whether a combination of low-dimensional chemical embeddings and a k-d tree data structure can achieve fast nearest neighbor queries while maintaining performance on standard chemical similarity search benchmarks. We examine different dimensionality reductions of standard chemical embeddings as well as a learned, structurally-aware embedding -- SmallSA -- for this task. With this framework, searches on over one billion chemicals execute in less than a second on a single CPU core, five orders of magnitude faster than the brute-force approach. We also demonstrate that SmallSA achieves competitive performance on chemical similarity benchmarks.
SALSA: Semantically-Aware Latent Space Autoencoder
Oct 04, 2023In deep learning for drug discovery, chemical data are often represented as simplified molecular-input line-entry system (SMILES) sequences which allow for straightforward implementation of natural language processing methodologies, one being the sequence-to-sequence autoencoder. However, we observe that training an autoencoder solely on SMILES is insufficient to learn molecular representations that are semantically meaningful, where semantics are defined by the structural (graph-to-graph) similarities between molecules. We demonstrate by example that autoencoders may map structurally similar molecules to distant codes, resulting in an incoherent latent space that does not respect the structural similarities between molecules. To address this shortcoming we propose Semantically-Aware Latent Space Autoencoder (SALSA), a transformer-autoencoder modified with a contrastive task, tailored specifically to learn graph-to-graph similarity between molecules. Formally, the contrastive objective is to map structurally similar molecules (separated by a single graph edit) to nearby codes in the latent space. To accomplish this, we generate a novel dataset comprised of sets of structurally similar molecules and opt for a supervised contrastive loss that is able to incorporate full sets of positive samples. We compare SALSA to its ablated counterparts, and show empirically that the composed training objective (reconstruction and contrastive task) leads to a higher quality latent space that is more 1) structurally-aware, 2) semantically continuous, and 3) property-aware.