 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeUtilizing Low-Dimensional Molecular Embeddings for Rapid Chemical Similarity Search
Feb 12, 2024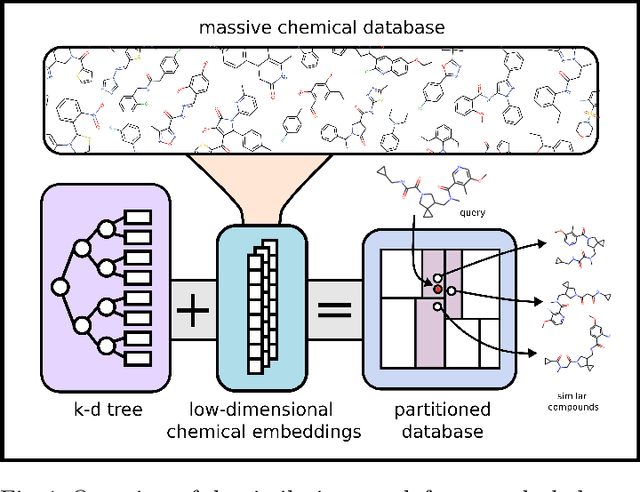
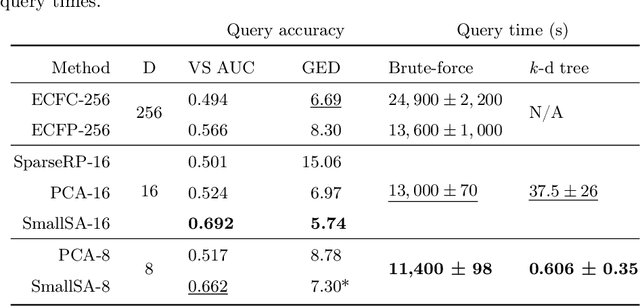
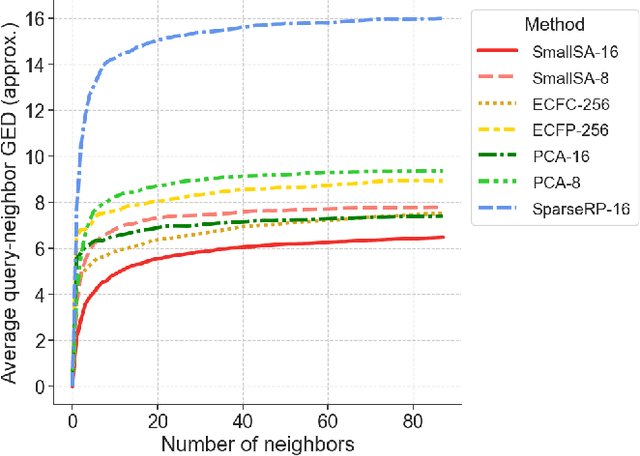
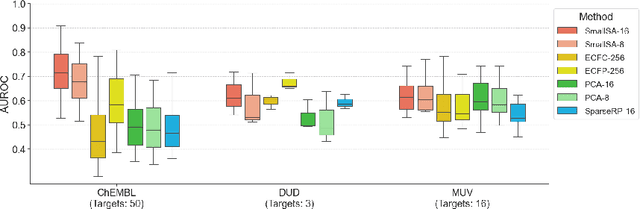
Nearest neighbor-based similarity searching is a common task in chemistry, with notable use cases in drug discovery. Yet, some of the most commonly used approaches for this task still leverage a brute-force approach. In practice this can be computationally costly and overly time-consuming, due in part to the sheer size of modern chemical databases. Previous computational advancements for this task have generally relied on improvements to hardware or dataset-specific tricks that lack generalizability. Approaches that leverage lower-complexity searching algorithms remain relatively underexplored. However, many of these algorithms are approximate solutions and/or struggle with typical high-dimensional chemical embeddings. Here we evaluate whether a combination of low-dimensional chemical embeddings and a k-d tree data structure can achieve fast nearest neighbor queries while maintaining performance on standard chemical similarity search benchmarks. We examine different dimensionality reductions of standard chemical embeddings as well as a learned, structurally-aware embedding -- SmallSA -- for this task. With this framework, searches on over one billion chemicals execute in less than a second on a single CPU core, five orders of magnitude faster than the brute-force approach. We also demonstrate that SmallSA achieves competitive performance on chemical similarity benchmarks.