 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeScaling Atomistic Protein Binder Design with Generative Pretraining and Test-Time Compute
Mar 30, 2026Protein interaction modeling is central to protein design, which has been transformed by machine learning with applications in drug discovery and beyond. In this landscape, structure-based de novo binder design is cast as either conditional generative modeling or sequence optimization via structure predictors ("hallucination"). We argue that this is a false dichotomy and propose Proteina-Complexa, a novel fully atomistic binder generation method unifying both paradigms. We extend recent flow-based latent protein generation architectures and leverage the domain-domain interactions of monomeric computationally predicted protein structures to construct Teddymer, a new large-scale dataset of synthetic binder-target pairs for pretraining. Combined with high-quality experimental multimers, this enables training a strong base model. We then perform inference-time optimization with this generative prior, unifying the strengths of previously distinct generative and hallucination methods. Proteina-Complexa sets a new state of the art in computational binder design benchmarks: it delivers markedly higher in-silico success rates than existing generative approaches, and our novel test-time optimization strategies greatly outperform previous hallucination methods under normalized compute budgets. We also demonstrate interface hydrogen bond optimization, fold class-guided binder generation, and extensions to small molecule targets and enzyme design tasks, again surpassing prior methods. Code, models and new data will be publicly released.
Rethinking Molecule Synthesizability with Chain-of-Reaction
Sep 19, 2025A well-known pitfall of molecular generative models is that they are not guaranteed to generate synthesizable molecules. There have been considerable attempts to address this problem, but given the exponentially large combinatorial space of synthesizable molecules, existing methods have shown limited coverage of the space and poor molecular optimization performance. To tackle these problems, we introduce ReaSyn, a generative framework for synthesizable projection where the model explores the neighborhood of given molecules in the synthesizable space by generating pathways that result in synthesizable analogs. To fully utilize the chemical knowledge contained in the synthetic pathways, we propose a novel perspective that views synthetic pathways akin to reasoning paths in large language models (LLMs). Specifically, inspired by chain-of-thought (CoT) reasoning in LLMs, we introduce the chain-of-reaction (CoR) notation that explicitly states reactants, reaction types, and intermediate products for each step in a pathway. With the CoR notation, ReaSyn can get dense supervision in every reaction step to explicitly learn chemical reaction rules during supervised training and perform step-by-step reasoning. In addition, to further enhance the reasoning capability of ReaSyn, we propose reinforcement learning (RL)-based finetuning and goal-directed test-time compute scaling tailored for synthesizable projection. ReaSyn achieves the highest reconstruction rate and pathway diversity in synthesizable molecule reconstruction and the highest optimization performance in synthesizable goal-directed molecular optimization, and significantly outperforms previous synthesizable projection methods in synthesizable hit expansion. These results highlight ReaSyn's superior ability to navigate combinatorially-large synthesizable chemical space.
Applications of Modular Co-Design for De Novo 3D Molecule Generation
May 23, 2025De novo 3D molecule generation is a pivotal task in drug discovery. However, many recent geometric generative models struggle to produce high-quality 3D structures, even if they maintain 2D validity and topological stability. To tackle this issue and enhance the learning of effective molecular generation dynamics, we present Megalodon-a family of scalable transformer models. These models are enhanced with basic equivariant layers and trained using a joint continuous and discrete denoising co-design objective. We assess Megalodon's performance on established molecule generation benchmarks and introduce new 3D structure benchmarks that evaluate a model's capability to generate realistic molecular structures, particularly focusing on energetics. We show that Megalodon achieves state-of-the-art results in 3D molecule generation, conditional structure generation, and structure energy benchmarks using diffusion and flow matching. Furthermore, doubling the number of parameters in Megalodon to 40M significantly enhances its performance, generating up to 49x more valid large molecules and achieving energy levels that are 2-10x lower than those of the best prior generative models.
GenMol: A Drug Discovery Generalist with Discrete Diffusion
Jan 10, 2025



Drug discovery is a complex process that involves multiple scenarios and stages, such as fragment-constrained molecule generation, hit generation and lead optimization. However, existing molecular generative models can only tackle one or two of these scenarios and lack the flexibility to address various aspects of the drug discovery pipeline. In this paper, we present Generalist Molecular generative model (GenMol), a versatile framework that addresses these limitations by applying discrete diffusion to the Sequential Attachment-based Fragment Embedding (SAFE) molecular representation. GenMol generates SAFE sequences through non-autoregressive bidirectional parallel decoding, thereby allowing utilization of a molecular context that does not rely on the specific token ordering and enhanced computational efficiency. Moreover, under the discrete diffusion framework, we introduce fragment remasking, a strategy that optimizes molecules by replacing fragments with masked tokens and regenerating them, enabling effective exploration of chemical space. GenMol significantly outperforms the previous GPT-based model trained on SAFE representations in de novo generation and fragment-constrained generation, and achieves state-of-the-art performance in goal-directed hit generation and lead optimization. These experimental results demonstrate that GenMol can tackle a wide range of drug discovery tasks, providing a unified and versatile approach for molecular design.
Molecule Generation with Fragment Retrieval Augmentation
Nov 18, 2024



Fragment-based drug discovery, in which molecular fragments are assembled into new molecules with desirable biochemical properties, has achieved great success. However, many fragment-based molecule generation methods show limited exploration beyond the existing fragments in the database as they only reassemble or slightly modify the given ones. To tackle this problem, we propose a new fragment-based molecule generation framework with retrieval augmentation, namely Fragment Retrieval-Augmented Generation (f-RAG). f-RAG is based on a pre-trained molecular generative model that proposes additional fragments from input fragments to complete and generate a new molecule. Given a fragment vocabulary, f-RAG retrieves two types of fragments: (1) hard fragments, which serve as building blocks that will be explicitly included in the newly generated molecule, and (2) soft fragments, which serve as reference to guide the generation of new fragments through a trainable fragment injection module. To extrapolate beyond the existing fragments, f-RAG updates the fragment vocabulary with generated fragments via an iterative refinement process which is further enhanced with post-hoc genetic fragment modification. f-RAG can achieve an improved exploration-exploitation trade-off by maintaining a pool of fragments and expanding it with novel and high-quality fragments through a strong generative prior.
BioNeMo Framework: a modular, high-performance library for AI model development in drug discovery
Nov 15, 2024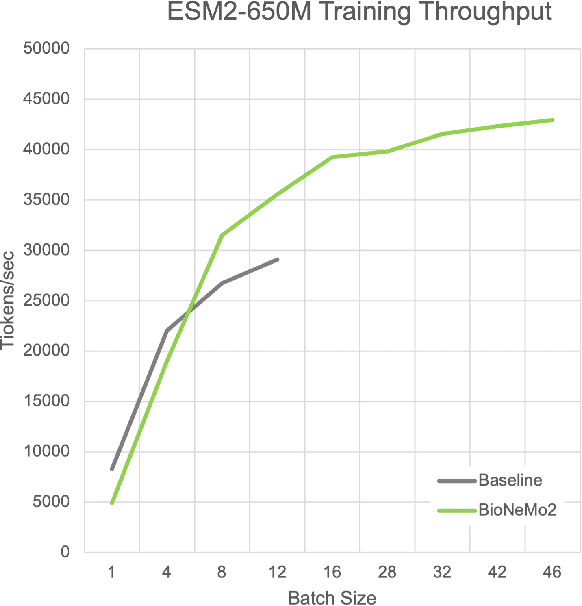
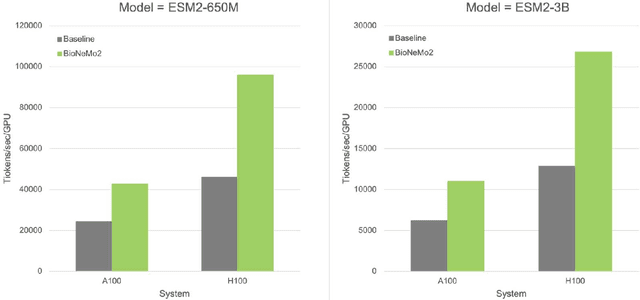
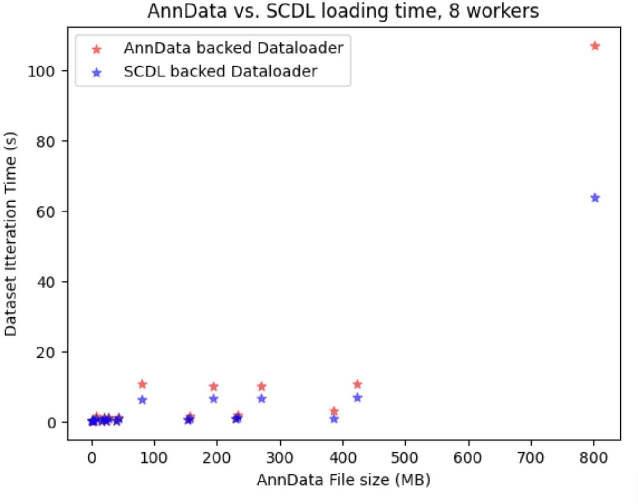
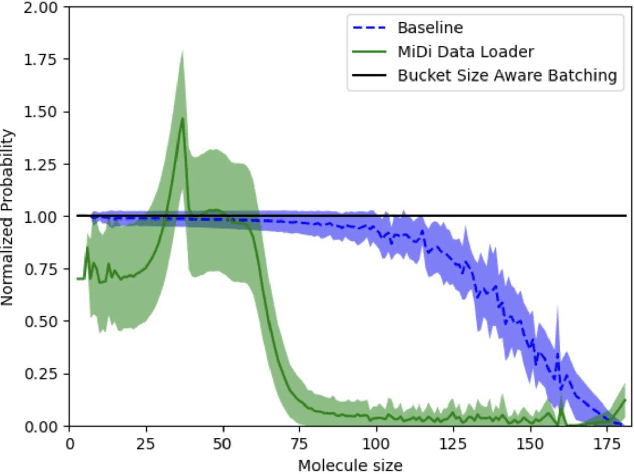
Artificial Intelligence models encoding biology and chemistry are opening new routes to high-throughput and high-quality in-silico drug development. However, their training increasingly relies on computational scale, with recent protein language models (pLM) training on hundreds of graphical processing units (GPUs). We introduce the BioNeMo Framework to facilitate the training of computational biology and chemistry AI models across hundreds of GPUs. Its modular design allows the integration of individual components, such as data loaders, into existing workflows and is open to community contributions. We detail technical features of the BioNeMo Framework through use cases such as pLM pre-training and fine-tuning. On 256 NVIDIA A100s, BioNeMo Framework trains a three billion parameter BERT-based pLM on over one trillion tokens in 4.2 days. The BioNeMo Framework is open-source and free for everyone to use.
General Binding Affinity Guidance for Diffusion Models in Structure-Based Drug Design
Jun 24, 2024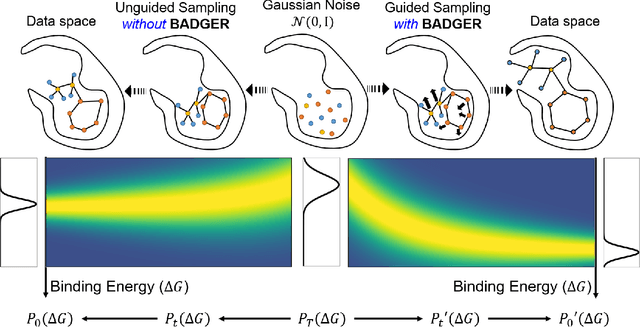
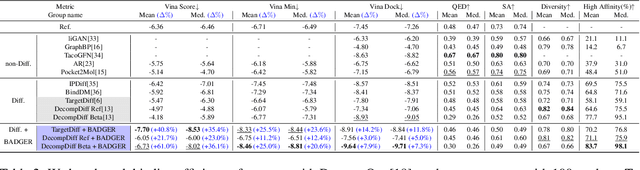
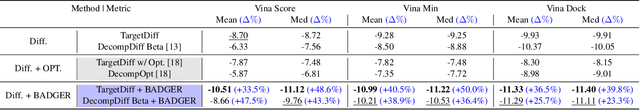
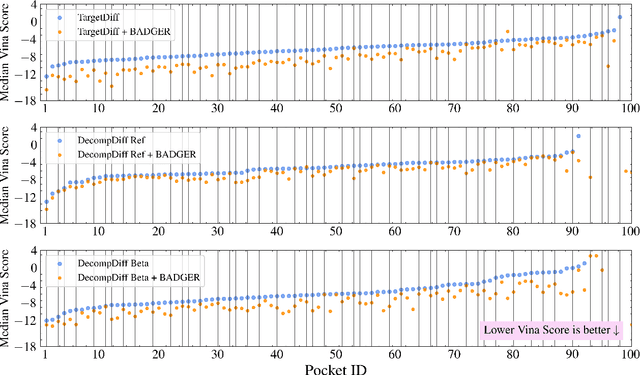
Structure-Based Drug Design (SBDD) focuses on generating valid ligands that strongly and specifically bind to a designated protein pocket. Several methods use machine learning for SBDD to generate these ligands in 3D space, conditioned on the structure of a desired protein pocket. Recently, diffusion models have shown success here by modeling the underlying distributions of atomic positions and types. While these methods are effective in considering the structural details of the protein pocket, they often fail to explicitly consider the binding affinity. Binding affinity characterizes how tightly the ligand binds to the protein pocket, and is measured by the change in free energy associated with the binding process. It is one of the most crucial metrics for benchmarking the effectiveness of the interaction between a ligand and protein pocket. To address this, we propose BADGER: Binding Affinity Diffusion Guidance with Enhanced Refinement. BADGER is a general guidance method to steer the diffusion sampling process towards improved protein-ligand binding, allowing us to adjust the distribution of the binding affinity between ligands and proteins. Our method is enabled by using a neural network (NN) to model the energy function, which is commonly approximated by AutoDock Vina (ADV). ADV's energy function is non-differentiable, and estimates the affinity based on the interactions between a ligand and target protein receptor. By using a NN as a differentiable energy function proxy, we utilize the gradient of our learned energy function as a guidance method on top of any trained diffusion model. We show that our method improves the binding affinity of generated ligands to their protein receptors by up to 60\%, significantly surpassing previous machine learning methods. We also show that our guidance method is flexible and can be easily applied to other diffusion-based SBDD frameworks.
CoarsenConf: Equivariant Coarsening with Aggregated Attention for Molecular Conformer Generation
Jun 26, 2023Molecular conformer generation (MCG) is an important task in cheminformatics and drug discovery. The ability to efficiently generate low-energy 3D structures can avoid expensive quantum mechanical simulations, leading to accelerated screenings and enhanced structural exploration. Several generative models have been developed for MCG, but many struggle to consistently produce high-quality conformers. To address these issues, we introduce CoarsenConf, which coarse-grains molecular graphs based on torsional angles and integrates them into an SE(3)-equivariant hierarchical variational autoencoder. Through equivariant coarse-graining, we aggregate the fine-grained atomic coordinates of subgraphs connected via rotatable bonds, creating a variable-length coarse-grained latent representation. Our model uses a novel aggregated attention mechanism to restore fine-grained coordinates from the coarse-grained latent representation, enabling efficient autoregressive generation of large molecules. Furthermore, our work expands current conformer generation benchmarks and introduces new metrics to better evaluate the quality and viability of generated conformers. We demonstrate that CoarsenConf generates more accurate conformer ensembles compared to prior generative models and traditional cheminformatics methods.
Improving Small Molecule Generation using Mutual Information Machine
Aug 18, 2022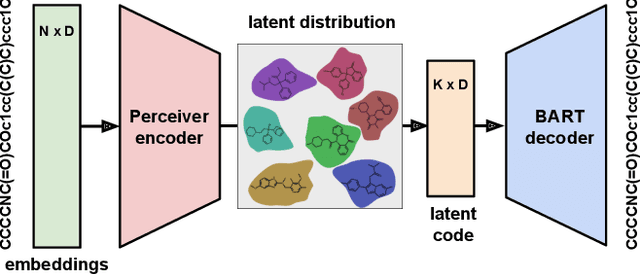
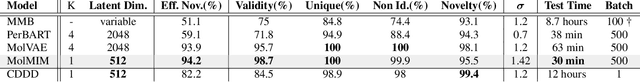
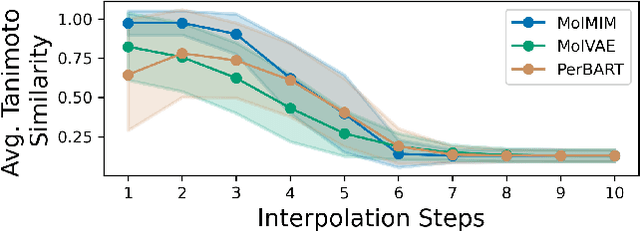
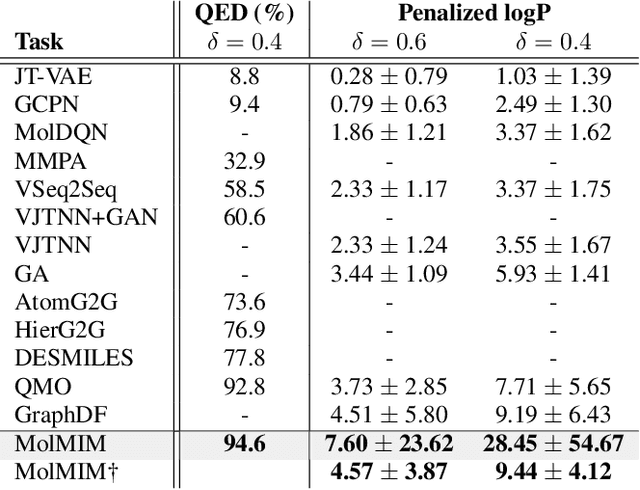
We address the task of controlled generation of small molecules, which entails finding novel molecules with desired properties under certain constraints (e.g., similarity to a reference molecule). Here we introduce MolMIM, a probabilistic auto-encoder for small molecule drug discovery that learns an informative and clustered latent space. MolMIM is trained with Mutual Information Machine (MIM) learning, and provides a fixed length representation of variable length SMILES strings. Since encoder-decoder models can learn representations with ``holes'' of invalid samples, here we propose a novel extension to the training procedure which promotes a dense latent space, and allows the model to sample valid molecules from random perturbations of latent codes. We provide a thorough comparison of MolMIM to several variable-size and fixed-size encoder-decoder models, demonstrating MolMIM's superior generation as measured in terms of validity, uniqueness, and novelty. We then utilize CMA-ES, a naive black-box and gradient free search algorithm, over MolMIM's latent space for the task of property guided molecule optimization. We achieve state-of-the-art results in several constrained single property optimization tasks as well as in the challenging task of multi-objective optimization, improving over previous success rate SOTA by more than 5\% . We attribute the strong results to MolMIM's latent representation which clusters similar molecules in the latent space, whereas CMA-ES is often used as a baseline optimization method. We also demonstrate MolMIM to be favourable in a compute limited regime, making it an attractive model for such cases.