 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeScalable Agentic Reasoning for Designing Biologics Targeting Intrinsically Disordered Proteins
Dec 17, 2025Intrinsically disordered proteins (IDPs) represent crucial therapeutic targets due to their significant role in disease -- approximately 80\% of cancer-related proteins contain long disordered regions -- but their lack of stable secondary/tertiary structures makes them "undruggable". While recent computational advances, such as diffusion models, can design high-affinity IDP binders, translating these to practical drug discovery requires autonomous systems capable of reasoning across complex conformational ensembles and orchestrating diverse computational tools at scale.To address this challenge, we designed and implemented StructBioReasoner, a scalable multi-agent system for designing biologics that can be used to target IDPs. StructBioReasoner employs a novel tournament-based reasoning framework where specialized agents compete to generate and refine therapeutic hypotheses, naturally distributing computational load for efficient exploration of the vast design space. Agents integrate domain knowledge with access to literature synthesis, AI-structure prediction, molecular simulations, and stability analysis, coordinating their execution on HPC infrastructure via an extensible federated agentic middleware, Academy. We benchmark StructBioReasoner across Der f 21 and NMNAT-2 and demonstrate that over 50\% of 787 designed and validated candidates for Der f 21 outperformed the human-designed reference binders from literature, in terms of improved binding free energy. For the more challenging NMNAT-2 protein, we identified three binding modes from 97,066 binders, including the well-studied NMNAT2:p53 interface. Thus, StructBioReasoner lays the groundwork for agentic reasoning systems for IDP therapeutic discovery on Exascale platforms.
Self Distillation Fine-Tuning of Protein Language Models Improves Versatility in Protein Design
Dec 10, 2025Supervised fine-tuning (SFT) is a standard approach for adapting large language models to specialized domains, yet its application to protein sequence modeling and protein language models (PLMs) remains ad hoc. This is in part because high-quality annotated data are far more difficult to obtain for proteins than for natural language. We present a simple and general recipe for fast SFT of PLMs, designed to improve the fidelity, reliability, and novelty of generated protein sequences. Unlike existing approaches that require costly precompiled experimental datasets for SFT, our method leverages the PLM itself, integrating a lightweight curation pipeline with domain-specific filters to construct high-quality training data. These filters can independently refine a PLM's output and identify candidates for in vitro evaluation; when combined with SFT, they enable PLMs to generate more stable and functional enzymes, while expanding exploration into protein sequence space beyond natural variants. Although our approach is agnostic to both the choice of protein language model (PLM) and the protein system, we demonstrate its effectiveness with a genome-scale PLM (GenSLM) applied to the tryptophan synthase enzyme family. The supervised fine-tuned model generates sequences that are not only more novel but also display improved characteristics across both targeted design constraints and emergent protein property measures.
HiPerRAG: High-Performance Retrieval Augmented Generation for Scientific Insights
May 07, 2025The volume of scientific literature is growing exponentially, leading to underutilized discoveries, duplicated efforts, and limited cross-disciplinary collaboration. Retrieval Augmented Generation (RAG) offers a way to assist scientists by improving the factuality of Large Language Models (LLMs) in processing this influx of information. However, scaling RAG to handle millions of articles introduces significant challenges, including the high computational costs associated with parsing documents and embedding scientific knowledge, as well as the algorithmic complexity of aligning these representations with the nuanced semantics of scientific content. To address these issues, we introduce HiPerRAG, a RAG workflow powered by high performance computing (HPC) to index and retrieve knowledge from more than 3.6 million scientific articles. At its core are Oreo, a high-throughput model for multimodal document parsing, and ColTrast, a query-aware encoder fine-tuning algorithm that enhances retrieval accuracy by using contrastive learning and late-interaction techniques. HiPerRAG delivers robust performance on existing scientific question answering benchmarks and two new benchmarks introduced in this work, achieving 90% accuracy on SciQ and 76% on PubMedQA-outperforming both domain-specific models like PubMedGPT and commercial LLMs such as GPT-4. Scaling to thousands of GPUs on the Polaris, Sunspot, and Frontier supercomputers, HiPerRAG delivers million document-scale RAG workflows for unifying scientific knowledge and fostering interdisciplinary innovation.
AdaParse: An Adaptive Parallel PDF Parsing and Resource Scaling Engine
Apr 23, 2025Language models for scientific tasks are trained on text from scientific publications, most distributed as PDFs that require parsing. PDF parsing approaches range from inexpensive heuristics (for simple documents) to computationally intensive ML-driven systems (for complex or degraded ones). The choice of the "best" parser for a particular document depends on its computational cost and the accuracy of its output. To address these issues, we introduce an Adaptive Parallel PDF Parsing and Resource Scaling Engine (AdaParse), a data-driven strategy for assigning an appropriate parser to each document. We enlist scientists to select preferred parser outputs and incorporate this information through direct preference optimization (DPO) into AdaParse, thereby aligning its selection process with human judgment. AdaParse then incorporates hardware requirements and predicted accuracy of each parser to orchestrate computational resources efficiently for large-scale parsing campaigns. We demonstrate that AdaParse, when compared to state-of-the-art parsers, improves throughput by $17\times$ while still achieving comparable accuracy (0.2 percent better) on a benchmark set of 1000 scientific documents. AdaParse's combination of high accuracy and parallel scalability makes it feasible to parse large-scale scientific document corpora to support the development of high-quality, trillion-token-scale text datasets. The implementation is available at https://github.com/7shoe/AdaParse/
ACE-RLHF: Automated Code Evaluation and Socratic Feedback Generation Tool using Large Language Models and Reinforcement Learning with Human Feedback
Apr 07, 2025Automated Program Repair tools are developed for generating feedback and suggesting a repair method for erroneous code. State of the art (SOTA) code repair methods rely on data-driven approaches and often fail to deliver solution for complicated programming questions. To interpret the natural language of unprecedented programming problems, using Large Language Models (LLMs) for code-feedback generation is crucial. LLMs generate more comprehensible feedback than compiler-generated error messages, and Reinforcement Learning with Human Feedback (RLHF) further enhances quality by integrating human-in-the-loop which helps novice students to lean programming from scratch interactively. We are applying RLHF fine-tuning technique for an expected Socratic response such as a question with hint to solve the programming issue. We are proposing code feedback generation tool by fine-tuning LLM with RLHF, Automated Code Evaluation with RLHF (ACE-RLHF), combining two open-source LLM models with two different SOTA optimization techniques. The quality of feedback is evaluated on two benchmark datasets containing basic and competition-level programming questions where the later is proposed by us. We achieved 2-5% higher accuracy than RL-free SOTA techniques using Llama-3-7B-Proximal-policy optimization in automated evaluation and similar or slightly higher accuracy compared to reward model-free RL with AI Feedback (RLAIF). We achieved almost 40% higher accuracy with GPT-3.5 Best-of-n optimization while performing manual evaluation.
DML-RAM: Deep Multimodal Learning Framework for Robotic Arm Manipulation using Pre-trained Models
Apr 04, 2025This paper presents a novel deep learning framework for robotic arm manipulation that integrates multimodal inputs using a late-fusion strategy. Unlike traditional end-to-end or reinforcement learning approaches, our method processes image sequences with pre-trained models and robot state data with machine learning algorithms, fusing their outputs to predict continuous action values for control. Evaluated on BridgeData V2 and Kuka datasets, the best configuration (VGG16 + Random Forest) achieved MSEs of 0.0021 and 0.0028, respectively, demonstrating strong predictive performance and robustness. The framework supports modularity, interpretability, and real-time decision-making, aligning with the goals of adaptive, human-in-the-loop cyber-physical systems.
MORAL: A Multimodal Reinforcement Learning Framework for Decision Making in Autonomous Laboratories
Apr 04, 2025We propose MORAL (a multimodal reinforcement learning framework for decision making in autonomous laboratories) that enhances sequential decision-making in autonomous robotic laboratories through the integration of visual and textual inputs. Using the BridgeData V2 dataset, we generate fine-tuned image captions with a pretrained BLIP-2 vision-language model and combine them with visual features through an early fusion strategy. The fused representations are processed using Deep Q-Network (DQN) and Proximal Policy Optimization (PPO) agents. Experimental results demonstrate that multimodal agents achieve a 20% improvement in task completion rates and significantly outperform visual-only and textual-only baselines after sufficient training. Compared to transformer-based and recurrent multimodal RL models, our approach achieves superior performance in cumulative reward and caption quality metrics (BLEU, METEOR, ROUGE-L). These results highlight the impact of semantically aligned language cues in enhancing agent learning efficiency and generalization. The proposed framework contributes to the advancement of multimodal reinforcement learning and embodied AI systems in dynamic, real-world environments.
Enhanced Penalty-based Bidirectional Reinforcement Learning Algorithms
Apr 04, 2025This research focuses on enhancing reinforcement learning (RL) algorithms by integrating penalty functions to guide agents in avoiding unwanted actions while optimizing rewards. The goal is to improve the learning process by ensuring that agents learn not only suitable actions but also which actions to avoid. Additionally, we reintroduce a bidirectional learning approach that enables agents to learn from both initial and terminal states, thereby improving speed and robustness in complex environments. Our proposed Penalty-Based Bidirectional methodology is tested against Mani skill benchmark environments, demonstrating an optimality improvement of success rate of approximately 4% compared to existing RL implementations. The findings indicate that this integrated strategy enhances policy learning, adaptability, and overall performance in challenging scenarios
Connecting Large Language Model Agent to High Performance Computing Resource
Feb 17, 2025The Large Language Model agent workflow enables the LLM to invoke tool functions to increase the performance on specific scientific domain questions. To tackle large scale of scientific research, it requires access to computing resource and parallel computing setup. In this work, we implemented Parsl to the LangChain/LangGraph tool call setup, to bridge the gap between the LLM agent to the computing resource. Two tool call implementations were set up and tested on both local workstation and HPC environment on Polaris/ALCF. The first implementation with Parsl-enabled LangChain tool node queues the tool functions concurrently to the Parsl workers for parallel execution. The second configuration is implemented by converting the tool functions into Parsl ensemble functions, and is more suitable for large task on super computer environment. The LLM agent workflow was prompted to run molecular dynamics simulations, with different protein structure and simulation conditions. These results showed the LLM agent tools were managed and executed concurrently by Parsl on the available computing resource.
BioNeMo Framework: a modular, high-performance library for AI model development in drug discovery
Nov 15, 2024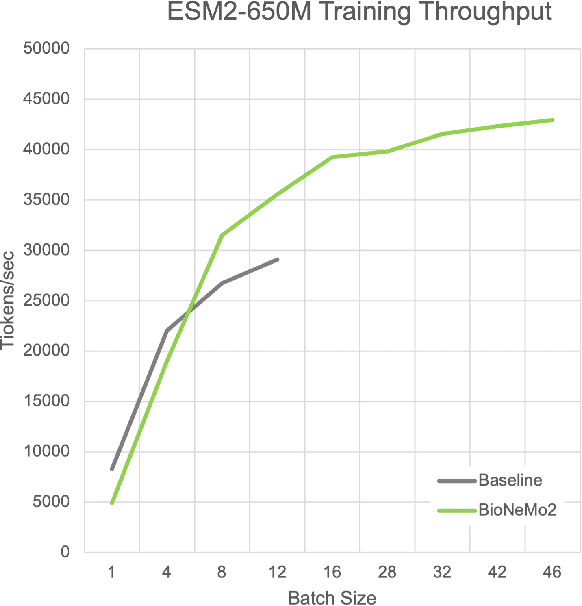
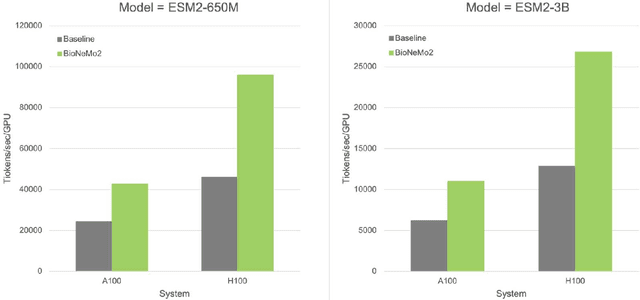
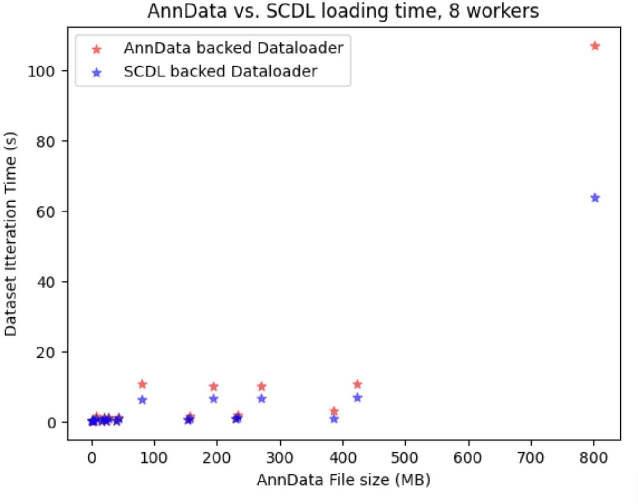
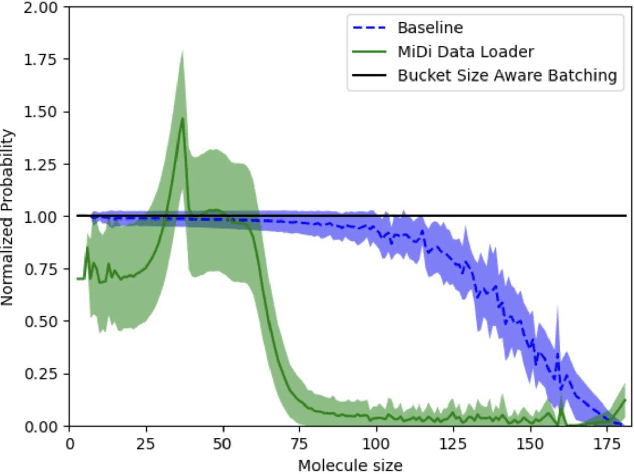
Artificial Intelligence models encoding biology and chemistry are opening new routes to high-throughput and high-quality in-silico drug development. However, their training increasingly relies on computational scale, with recent protein language models (pLM) training on hundreds of graphical processing units (GPUs). We introduce the BioNeMo Framework to facilitate the training of computational biology and chemistry AI models across hundreds of GPUs. Its modular design allows the integration of individual components, such as data loaders, into existing workflows and is open to community contributions. We detail technical features of the BioNeMo Framework through use cases such as pLM pre-training and fine-tuning. On 256 NVIDIA A100s, BioNeMo Framework trains a three billion parameter BERT-based pLM on over one trillion tokens in 4.2 days. The BioNeMo Framework is open-source and free for everyone to use.