 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeScalable Agentic Reasoning for Designing Biologics Targeting Intrinsically Disordered Proteins
Dec 17, 2025Intrinsically disordered proteins (IDPs) represent crucial therapeutic targets due to their significant role in disease -- approximately 80\% of cancer-related proteins contain long disordered regions -- but their lack of stable secondary/tertiary structures makes them "undruggable". While recent computational advances, such as diffusion models, can design high-affinity IDP binders, translating these to practical drug discovery requires autonomous systems capable of reasoning across complex conformational ensembles and orchestrating diverse computational tools at scale.To address this challenge, we designed and implemented StructBioReasoner, a scalable multi-agent system for designing biologics that can be used to target IDPs. StructBioReasoner employs a novel tournament-based reasoning framework where specialized agents compete to generate and refine therapeutic hypotheses, naturally distributing computational load for efficient exploration of the vast design space. Agents integrate domain knowledge with access to literature synthesis, AI-structure prediction, molecular simulations, and stability analysis, coordinating their execution on HPC infrastructure via an extensible federated agentic middleware, Academy. We benchmark StructBioReasoner across Der f 21 and NMNAT-2 and demonstrate that over 50\% of 787 designed and validated candidates for Der f 21 outperformed the human-designed reference binders from literature, in terms of improved binding free energy. For the more challenging NMNAT-2 protein, we identified three binding modes from 97,066 binders, including the well-studied NMNAT2:p53 interface. Thus, StructBioReasoner lays the groundwork for agentic reasoning systems for IDP therapeutic discovery on Exascale platforms.
HiPerRAG: High-Performance Retrieval Augmented Generation for Scientific Insights
May 07, 2025The volume of scientific literature is growing exponentially, leading to underutilized discoveries, duplicated efforts, and limited cross-disciplinary collaboration. Retrieval Augmented Generation (RAG) offers a way to assist scientists by improving the factuality of Large Language Models (LLMs) in processing this influx of information. However, scaling RAG to handle millions of articles introduces significant challenges, including the high computational costs associated with parsing documents and embedding scientific knowledge, as well as the algorithmic complexity of aligning these representations with the nuanced semantics of scientific content. To address these issues, we introduce HiPerRAG, a RAG workflow powered by high performance computing (HPC) to index and retrieve knowledge from more than 3.6 million scientific articles. At its core are Oreo, a high-throughput model for multimodal document parsing, and ColTrast, a query-aware encoder fine-tuning algorithm that enhances retrieval accuracy by using contrastive learning and late-interaction techniques. HiPerRAG delivers robust performance on existing scientific question answering benchmarks and two new benchmarks introduced in this work, achieving 90% accuracy on SciQ and 76% on PubMedQA-outperforming both domain-specific models like PubMedGPT and commercial LLMs such as GPT-4. Scaling to thousands of GPUs on the Polaris, Sunspot, and Frontier supercomputers, HiPerRAG delivers million document-scale RAG workflows for unifying scientific knowledge and fostering interdisciplinary innovation.
Connecting Large Language Model Agent to High Performance Computing Resource
Feb 17, 2025The Large Language Model agent workflow enables the LLM to invoke tool functions to increase the performance on specific scientific domain questions. To tackle large scale of scientific research, it requires access to computing resource and parallel computing setup. In this work, we implemented Parsl to the LangChain/LangGraph tool call setup, to bridge the gap between the LLM agent to the computing resource. Two tool call implementations were set up and tested on both local workstation and HPC environment on Polaris/ALCF. The first implementation with Parsl-enabled LangChain tool node queues the tool functions concurrently to the Parsl workers for parallel execution. The second configuration is implemented by converting the tool functions into Parsl ensemble functions, and is more suitable for large task on super computer environment. The LLM agent workflow was prompted to run molecular dynamics simulations, with different protein structure and simulation conditions. These results showed the LLM agent tools were managed and executed concurrently by Parsl on the available computing resource.
DeepSpeed4Science Initiative: Enabling Large-Scale Scientific Discovery through Sophisticated AI System Technologies
Oct 11, 2023



In the upcoming decade, deep learning may revolutionize the natural sciences, enhancing our capacity to model and predict natural occurrences. This could herald a new era of scientific exploration, bringing significant advancements across sectors from drug development to renewable energy. To answer this call, we present DeepSpeed4Science initiative (deepspeed4science.ai) which aims to build unique capabilities through AI system technology innovations to help domain experts to unlock today's biggest science mysteries. By leveraging DeepSpeed's current technology pillars (training, inference and compression) as base technology enablers, DeepSpeed4Science will create a new set of AI system technologies tailored for accelerating scientific discoveries by addressing their unique complexity beyond the common technical approaches used for accelerating generic large language models (LLMs). In this paper, we showcase the early progress we made with DeepSpeed4Science in addressing two of the critical system challenges in structural biology research.
Achieving 100X faster simulations of complex biological phenomena by coupling ML to HPC ensembles
Apr 26, 2021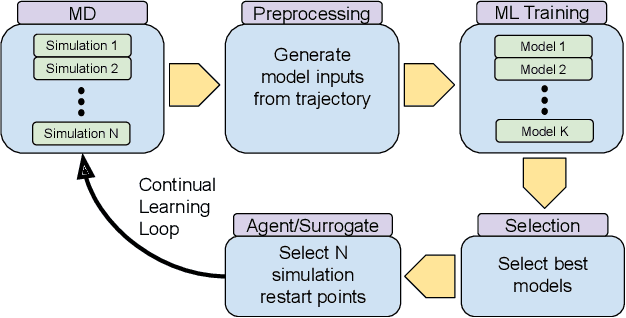
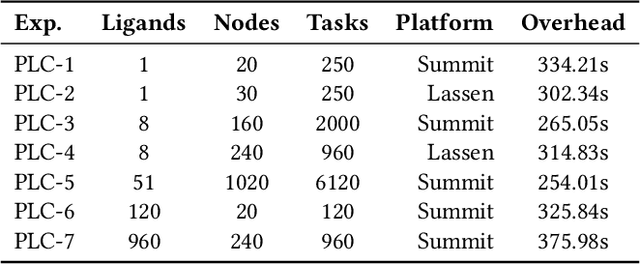
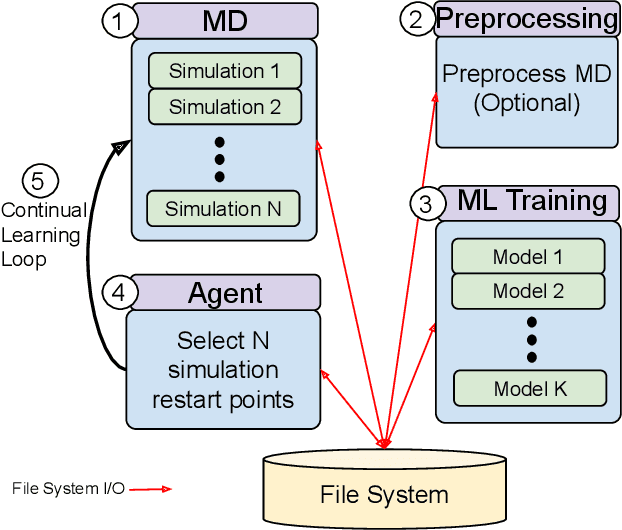
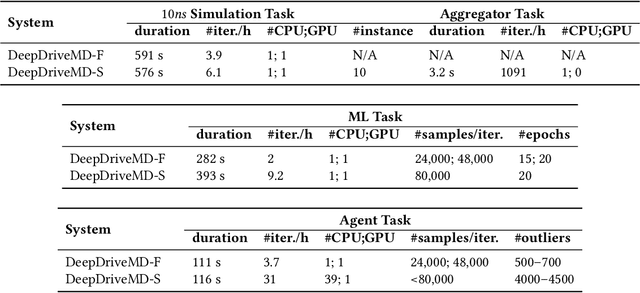
The use of ML methods to dynamically steer ensemble-based simulations promises significant improvements in the performance of scientific applications. We present DeepDriveMD, a tool for a range of prototypical ML-driven HPC simulation scenarios, and use it to quantify improvements in the scientific performance of ML-driven ensemble-based applications. We discuss its design and characterize its performance. Motivated by the potential for further scientific improvements and applicability to more sophisticated physical systems, we extend the design of DeepDriveMD to support stream-based communication between simulations and learning methods. It demonstrates a 100x speedup to fold proteins, and performs 1.6x more simulations per unit time, improving resource utilization compared to the sequential framework. Experiments are performed on leadership-class platforms, at scales of up to O(1000) nodes, and for production workloads. We establish DeepDriveMD as a high-performance framework for ML-driven HPC simulation scenarios, that supports diverse simulation and ML back-ends, and which enables new scientific insights by improving length- and time-scale accessed.
Pandemic Drugs at Pandemic Speed: Accelerating COVID-19 Drug Discovery with Hybrid Machine Learning- and Physics-based Simulations on High Performance Computers
Mar 04, 2021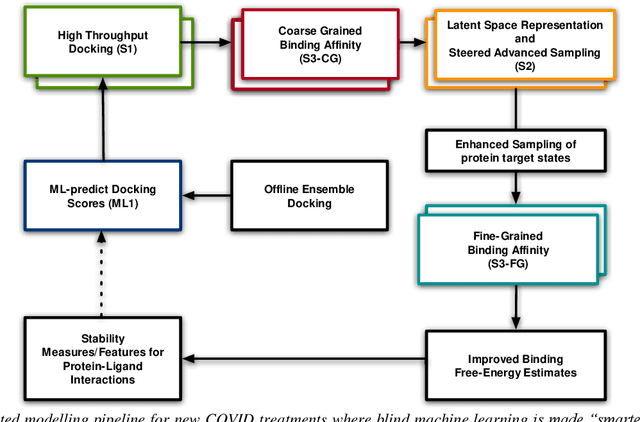

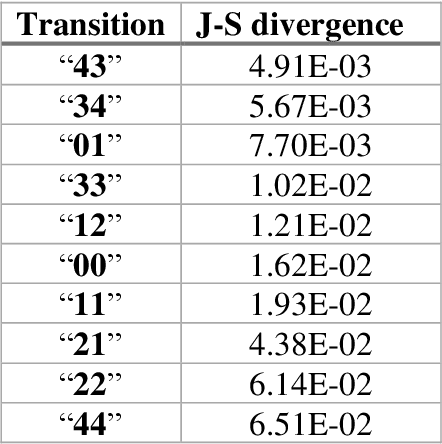
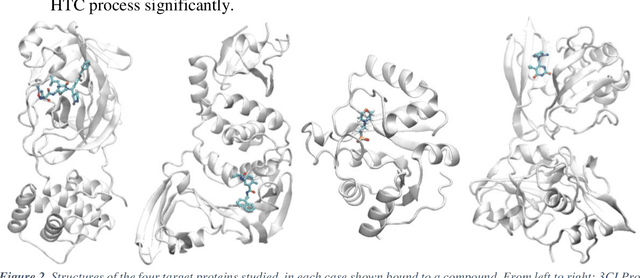
The race to meet the challenges of the global pandemic has served as a reminder that the existing drug discovery process is expensive, inefficient and slow. There is a major bottleneck screening the vast number of potential small molecules to shortlist lead compounds for antiviral drug development. New opportunities to accelerate drug discovery lie at the interface between machine learning methods, in this case developed for linear accelerators, and physics-based methods. The two in silico methods, each have their own advantages and limitations which, interestingly, complement each other. Here, we present an innovative method that combines both approaches to accelerate drug discovery. The scale of the resulting workflow is such that it is dependent on high performance computing. We have demonstrated the applicability of this workflow on four COVID-19 target proteins and our ability to perform the required large-scale calculations to identify lead compounds on a variety of supercomputers.
Artificial intelligence techniques for integrative structural biology of intrinsically disordered proteins
Dec 01, 2020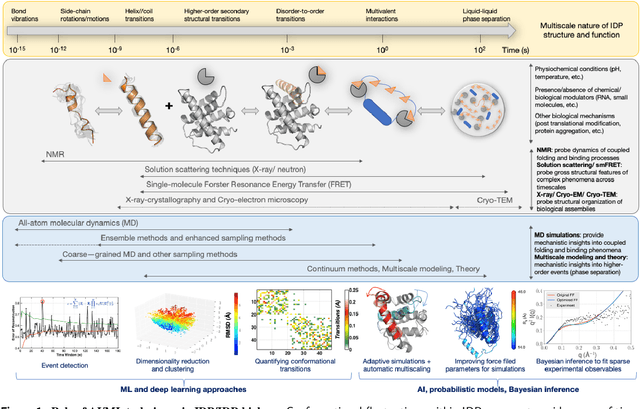
We outline recent developments in artificial intelligence (AI) and machine learning (ML) techniques for integrative structural biology of intrinsically disordered proteins (IDP) ensembles. IDPs challenge the traditional protein structure-function paradigm by adapting their conformations in response to specific binding partners leading them to mediate diverse, and often complex cellular functions such as biological signaling, self organization and compartmentalization. Obtaining mechanistic insights into their function can therefore be challenging for traditional structural determination techniques. Often, scientists have to rely on piecemeal evidence drawn from diverse experimental techniques to characterize their functional mechanisms. Multiscale simulations can help bridge critical knowledge gaps about IDP structure function relationships - however, these techniques also face challenges in resolving emergent phenomena within IDP conformational ensembles. We posit that scalable statistical inference techniques can effectively integrate information gleaned from multiple experimental techniques as well as from simulations, thus providing access to atomistic details of these emergent phenomena.
DeepDriveMD: Deep-Learning Driven Adaptive Molecular Simulations for Protein Folding
Sep 17, 2019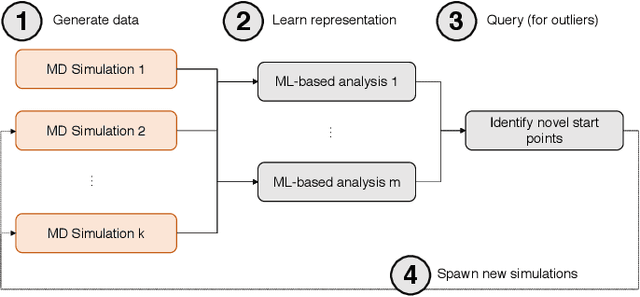
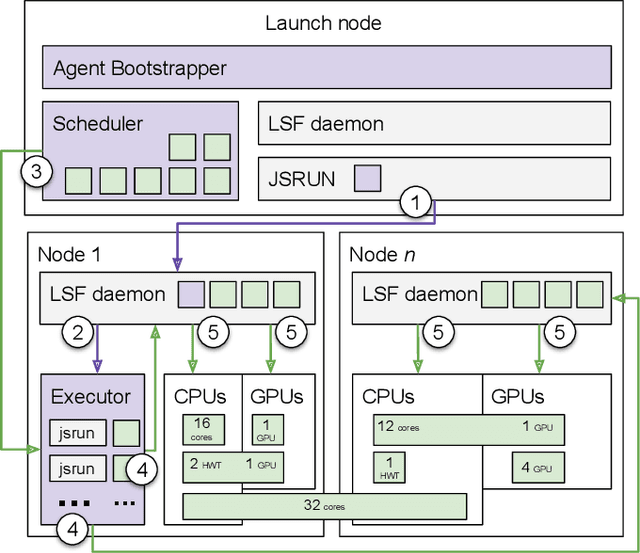
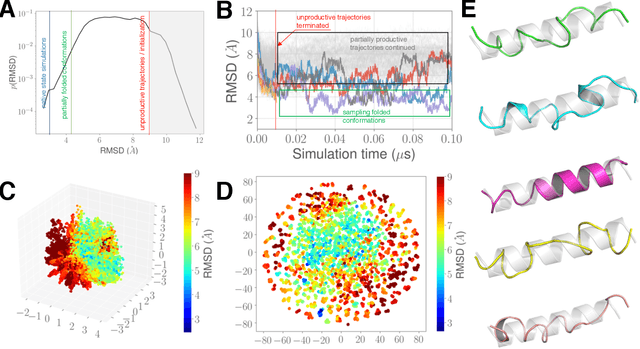
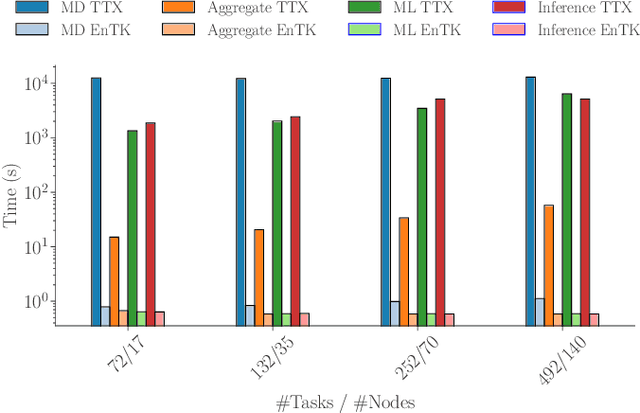
Simulations of biological macromolecules play an important role in understanding the physical basis of a number of complex processes such as protein folding. Even with increasing computational power and evolution of specialized architectures, the ability to simulate protein folding at atomistic scales still remains challenging. This stems from the dual aspects of high dimensionality of protein conformational landscapes, and the inability of atomistic molecular dynamics (MD) simulations to sufficiently sample these landscapes to observe folding events. Machine learning/deep learning (ML/DL) techniques, when combined with atomistic MD simulations offer the opportunity to potentially overcome these limitations by: (1) effectively reducing the dimensionality of MD simulations to automatically build latent representations that correspond to biophysically relevant reaction coordinates (RCs), and (2) driving MD simulations to automatically sample potentially novel conformational states based on these RCs. We examine how coupling DL approaches with MD simulations can fold small proteins effectively on supercomputers. In particular, we study the computational costs and effectiveness of scaling DL-coupled MD workflows by folding two prototypical systems, viz., Fs-peptide and the fast-folding variant of the villin head piece protein. We demonstrate that a DL driven MD workflow is able to effectively learn latent representations and drive adaptive simulations. Compared to traditional MD-based approaches, our approach achieves an effective performance gain in sampling the folded states by at least 2.3x. Our study provides a quantitative basis to understand how DL driven MD simulations, can lead to effective performance gains and reduced times to solution on supercomputing resources.