 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeImproved Dimensionality Reduction for Inverse Problems in Nuclear Fusion and High-Energy Astrophysics
May 05, 2025Many inverse problems in nuclear fusion and high-energy astrophysics research, such as the optimization of tokamak reactor geometries or the inference of black hole parameters from interferometric images, necessitate high-dimensional parameter scans and large ensembles of simulations to be performed. Such inverse problems typically involve large uncertainties, both in the measurement parameters being inverted and in the underlying physics models themselves. Monte Carlo sampling, when combined with modern non-linear dimensionality reduction techniques such as autoencoders and manifold learning, can be used to reduce the size of the parameter spaces considerably. However, there is no guarantee that the resulting combinations of parameters will be physically valid, or even mathematically consistent. In this position paper, we advocate adopting a hybrid approach that leverages our recent advances in the development of formal verification methods for numerical algorithms, with the goal of constructing parameter space restrictions with provable mathematical and physical correctness properties, whilst nevertheless respecting both experimental uncertainties and uncertainties in the underlying physical processes.
Scalable Runtime Architecture for Data-driven, Hybrid HPC and ML Workflow Applications
Mar 17, 2025Hybrid workflows combining traditional HPC and novel ML methodologies are transforming scientific computing. This paper presents the architecture and implementation of a scalable runtime system that extends RADICAL-Pilot with service-based execution to support AI-out-HPC workflows. Our runtime system enables distributed ML capabilities, efficient resource management, and seamless HPC/ML coupling across local and remote platforms. Preliminary experimental results show that our approach manages concurrent execution of ML models across local and remote HPC/cloud resources with minimal architectural overheads. This lays the foundation for prototyping three representative data-driven workflow applications and executing them at scale on leadership-class HPC platforms.
Envisioning National Resources for Artificial Intelligence Research: NSF Workshop Report
Dec 13, 2024This is a report of an NSF workshop titled "Envisioning National Resources for Artificial Intelligence Research" held in Alexandria, Virginia, in May 2024. The workshop aimed to identify initial challenges and opportunities for national resources for AI research (e.g., compute, data, models, etc.) and to facilitate planning for the envisioned National AI Research Resource. Participants included AI and cyberinfrastructure (CI) experts. The report outlines significant findings and identifies needs and recommendations from the workshop.
Feasibility Study on Active Learning of Smart Surrogates for Scientific Simulations
Jul 10, 2024
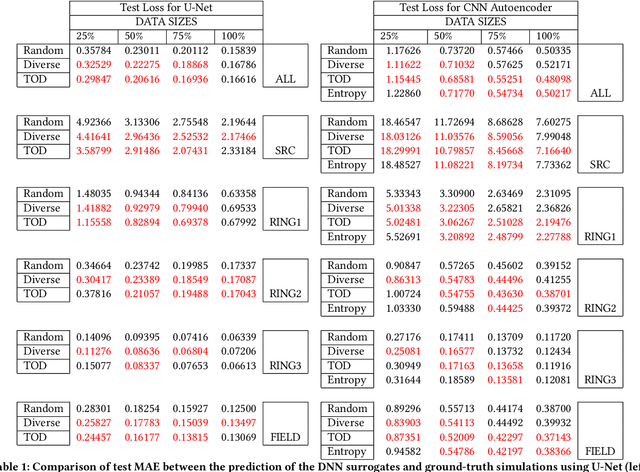


High-performance scientific simulations, important for comprehension of complex systems, encounter computational challenges especially when exploring extensive parameter spaces. There has been an increasing interest in developing deep neural networks (DNNs) as surrogate models capable of accelerating the simulations. However, existing approaches for training these DNN surrogates rely on extensive simulation data which are heuristically selected and generated with expensive computation -- a challenge under-explored in the literature. In this paper, we investigate the potential of incorporating active learning into DNN surrogate training. This allows intelligent and objective selection of training simulations, reducing the need to generate extensive simulation data as well as the dependency of the performance of DNN surrogates on pre-defined training simulations. In the problem context of constructing DNN surrogates for diffusion equations with sources, we examine the efficacy of diversity- and uncertainty-based strategies for selecting training simulations, considering two different DNN architecture. The results set the groundwork for developing the high-performance computing infrastructure for Smart Surrogates that supports on-the-fly generation of simulation data steered by active learning strategies to potentially improve the efficiency of scientific simulations.
AI-coupled HPC Workflows
Aug 24, 2022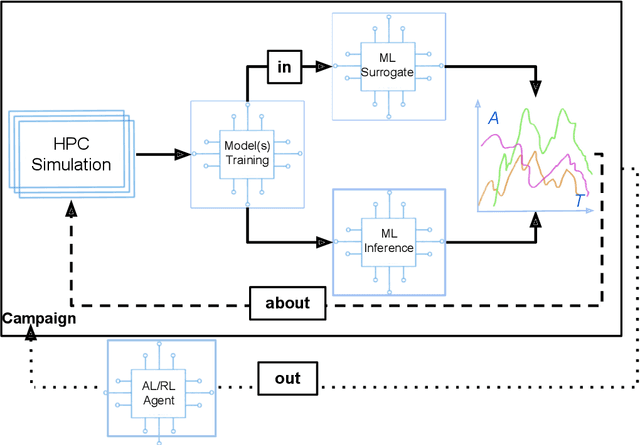
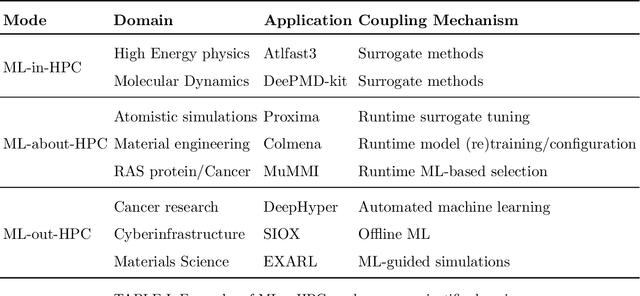
Increasingly, scientific discovery requires sophisticated and scalable workflows. Workflows have become the ``new applications,'' wherein multi-scale computing campaigns comprise multiple and heterogeneous executable tasks. In particular, the introduction of AI/ML models into the traditional HPC workflows has been an enabler of highly accurate modeling, typically reducing computational needs compared to traditional methods. This chapter discusses various modes of integrating AI/ML models to HPC computations, resulting in diverse types of AI-coupled HPC workflows. The increasing need of coupling AI/ML and HPC across scientific domains is motivated, and then exemplified by a number of production-grade use cases for each mode. We additionally discuss the primary challenges of extreme-scale AI-coupled HPC campaigns -- task heterogeneity, adaptivity, performance -- and several framework and middleware solutions which aim to address them. While both HPC workflow and AI/ML computing paradigms are independently effective, we highlight how their integration, and ultimate convergence, is leading to significant improvements in scientific performance across a range of domains, ultimately resulting in scientific explorations otherwise unattainable.
Asynchronous Execution of Heterogeneous Tasks in AI-coupled HPC Workflows
Aug 23, 2022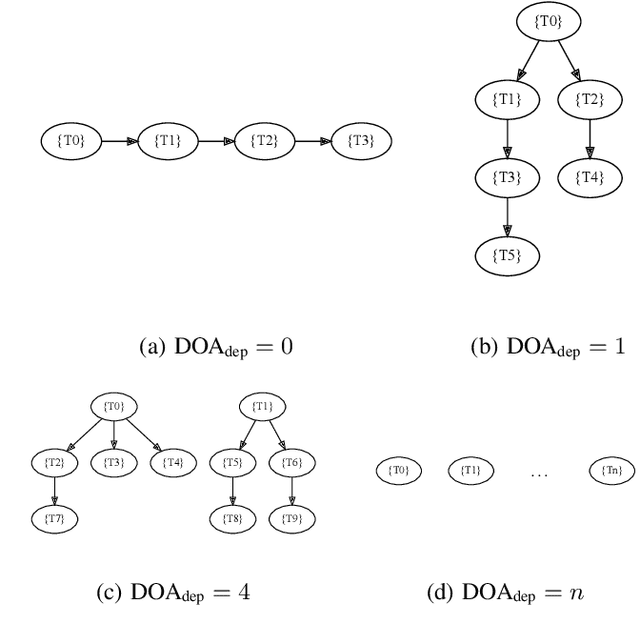
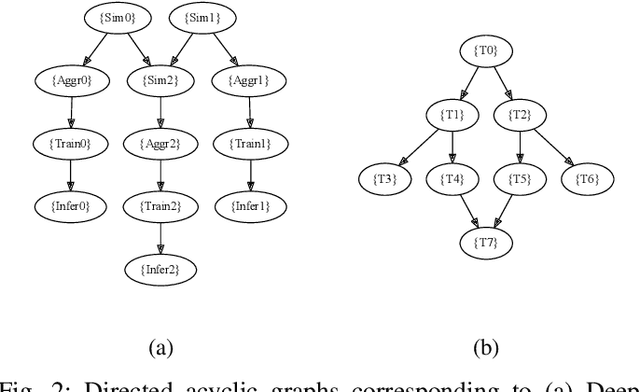
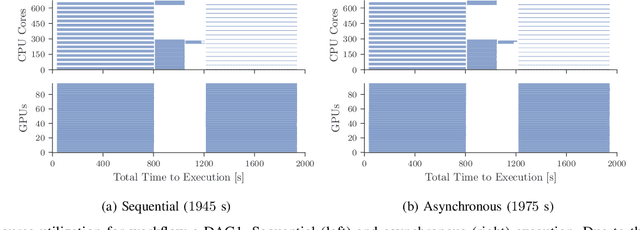
Heterogeneous scientific workflows consist of numerous types of tasks and dependencies between them. Middleware capable of scheduling and submitting different task types across heterogeneous platforms must permit asynchronous execution of tasks for improved resource utilization, task throughput, and reduced makespan. In this paper we present an analysis of an important class of heterogeneous workflows, viz., AI-driven HPC workflows, to investigate asynchronous task execution requirements and properties. We model the degree of asynchronicity permitted for arbitrary workflows, and propose key metrics that can be used to determine qualitative benefits when employing asynchronous execution. Our experiments represent important scientific drivers, are performed at scale on Summit, and performance enhancements due to asynchronous execution are consistent with our model.
Optimal Decision Making in High-Throughput Virtual Screening Pipelines
Sep 23, 2021
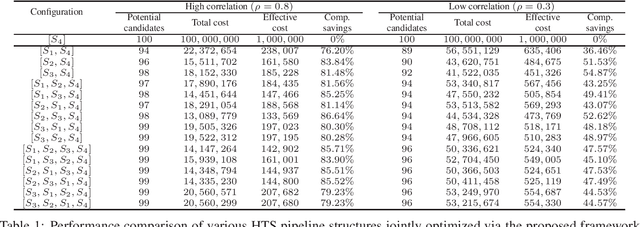
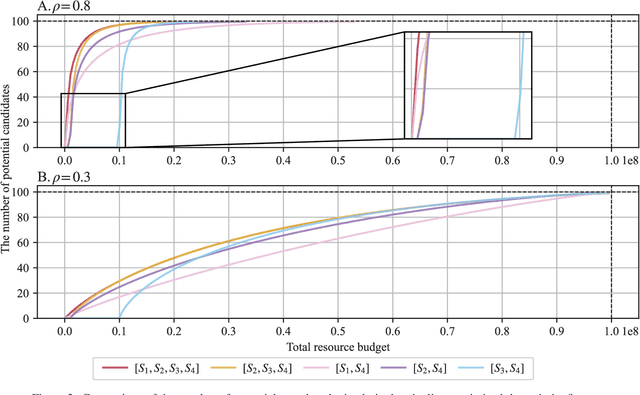
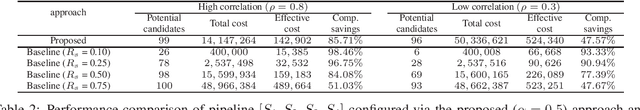
Effective selection of the potential candidates that meet certain conditions in a tremendously large search space has been one of the major concerns in many real-world applications. In addition to the nearly infinitely large search space, rigorous evaluation of a sample based on the reliable experimental or computational platform is often prohibitively expensive, making the screening problem more challenging. In such a case, constructing a high-throughput screening (HTS) pipeline that pre-sifts the samples expected to be potential candidates through the efficient earlier stages, results in a significant amount of savings in resources. However, to the best of our knowledge, despite many successful applications, no one has studied optimal pipeline design or optimal pipeline operations. In this study, we propose two optimization frameworks, applying to most (if not all) screening campaigns involving experimental or/and computational evaluations, for optimally determining the screening thresholds of an HTS pipeline. We validate the proposed frameworks on both analytic and practical scenarios. In particular, we consider the optimal computational campaign for the long non-coding RNA (lncRNA) classification as a practical example. To accomplish this, we built the high-throughput virtual screening (HTVS) pipeline for classifying the lncRNA. The simulation results demonstrate that the proposed frameworks significantly reduce the effective selection cost per potential candidate and make the HTS pipelines less sensitive to their structural variations. In addition to the validation, we provide insights on constructing a better HTS pipeline based on the simulation results.
Protein-Ligand Docking Surrogate Models: A SARS-CoV-2 Benchmark for Deep Learning Accelerated Virtual Screening
Jun 30, 2021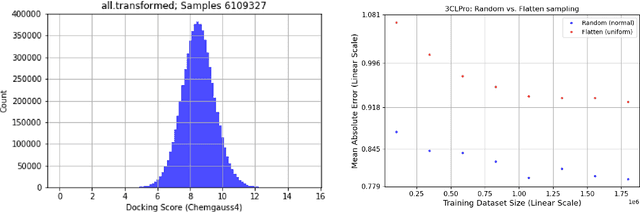

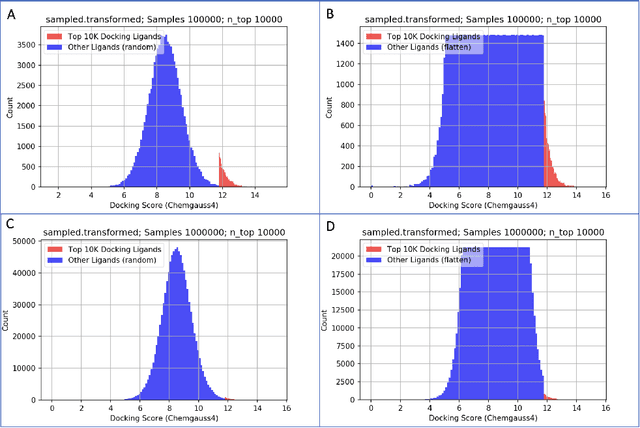
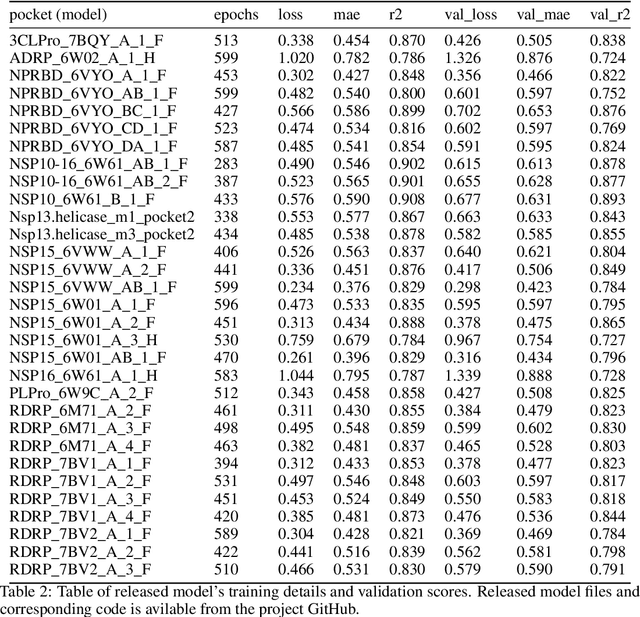
We propose a benchmark to study surrogate model accuracy for protein-ligand docking. We share a dataset consisting of 200 million 3D complex structures and 2D structure scores across a consistent set of 13 million "in-stock" molecules over 15 receptors, or binding sites, across the SARS-CoV-2 proteome. Our work shows surrogate docking models have six orders of magnitude more throughput than standard docking protocols on the same supercomputer node types. We demonstrate the power of high-speed surrogate models by running each target against 1 billion molecules in under a day (50k predictions per GPU seconds). We showcase a workflow for docking utilizing surrogate ML models as a pre-filter. Our workflow is ten times faster at screening a library of compounds than the standard technique, with an error rate less than 0.01\% of detecting the underlying best scoring 0.1\% of compounds. Our analysis of the speedup explains that to screen more molecules under a docking paradigm, another order of magnitude speedup must come from model accuracy rather than computing speed (which, if increased, will not anymore alter our throughput to screen molecules). We believe this is strong evidence for the community to begin focusing on improving the accuracy of surrogate models to improve the ability to screen massive compound libraries 100x or even 1000x faster than current techniques.
Achieving 100X faster simulations of complex biological phenomena by coupling ML to HPC ensembles
Apr 26, 2021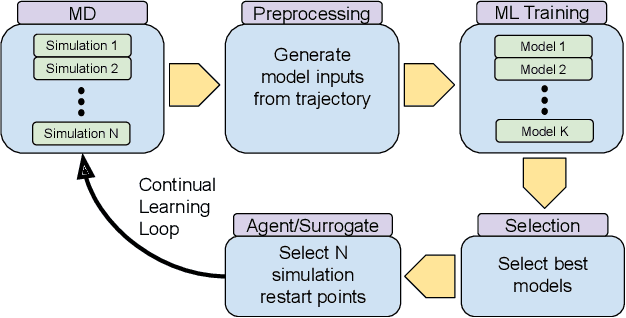
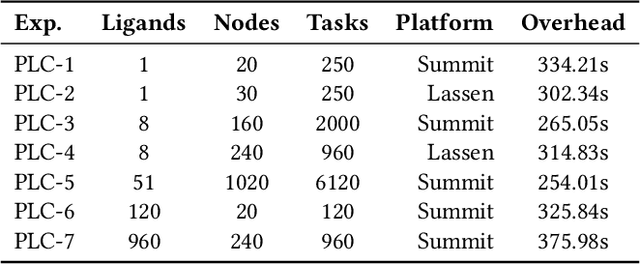
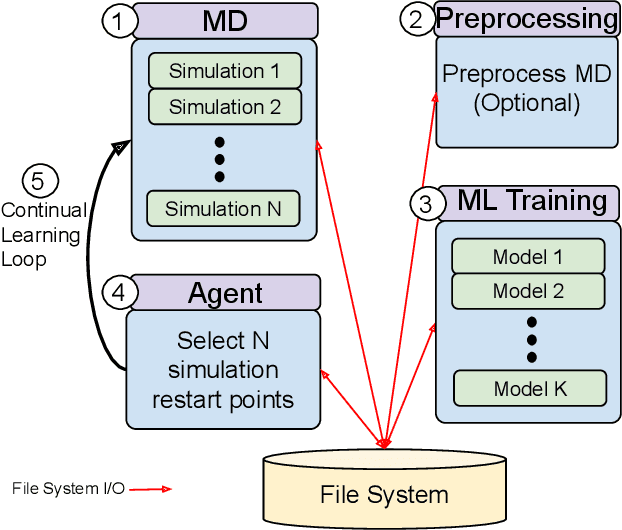
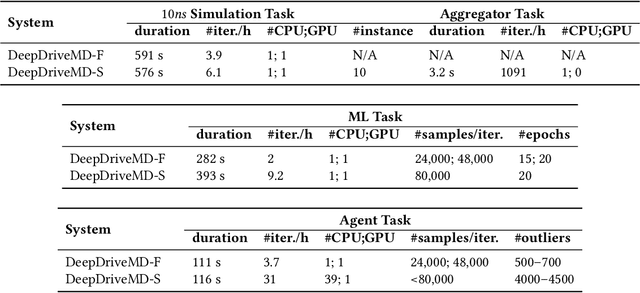
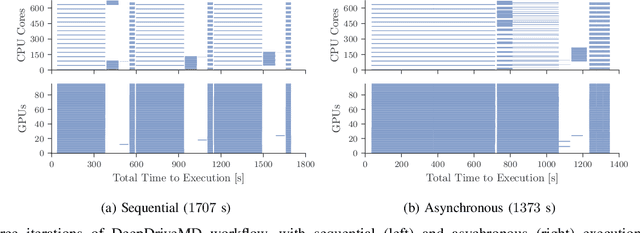
The use of ML methods to dynamically steer ensemble-based simulations promises significant improvements in the performance of scientific applications. We present DeepDriveMD, a tool for a range of prototypical ML-driven HPC simulation scenarios, and use it to quantify improvements in the scientific performance of ML-driven ensemble-based applications. We discuss its design and characterize its performance. Motivated by the potential for further scientific improvements and applicability to more sophisticated physical systems, we extend the design of DeepDriveMD to support stream-based communication between simulations and learning methods. It demonstrates a 100x speedup to fold proteins, and performs 1.6x more simulations per unit time, improving resource utilization compared to the sequential framework. Experiments are performed on leadership-class platforms, at scales of up to O(1000) nodes, and for production workloads. We establish DeepDriveMD as a high-performance framework for ML-driven HPC simulation scenarios, that supports diverse simulation and ML back-ends, and which enables new scientific insights by improving length- and time-scale accessed.
Pandemic Drugs at Pandemic Speed: Accelerating COVID-19 Drug Discovery with Hybrid Machine Learning- and Physics-based Simulations on High Performance Computers
Mar 04, 2021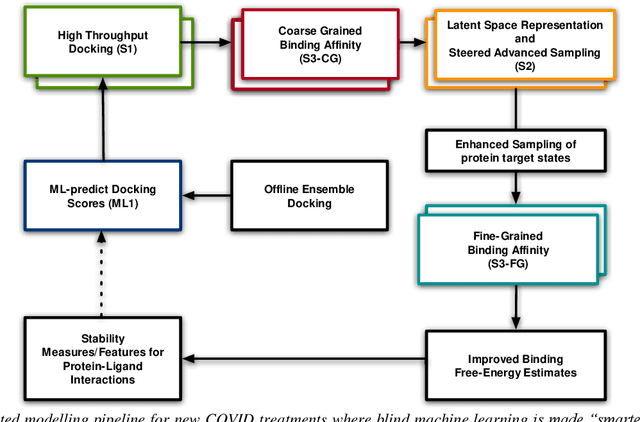

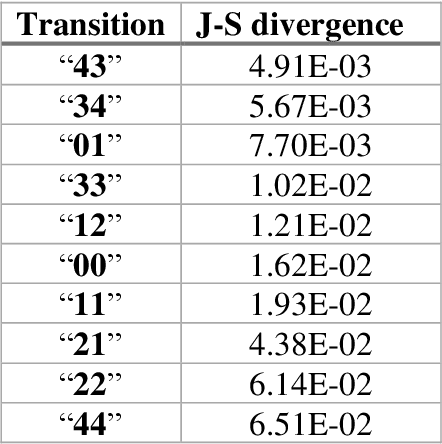
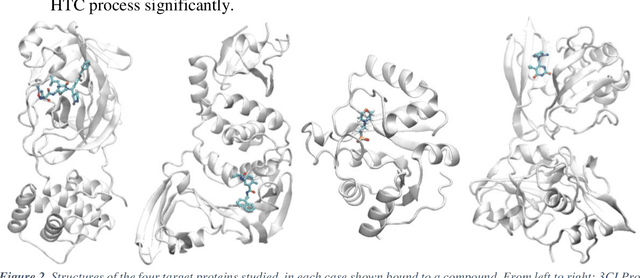
The race to meet the challenges of the global pandemic has served as a reminder that the existing drug discovery process is expensive, inefficient and slow. There is a major bottleneck screening the vast number of potential small molecules to shortlist lead compounds for antiviral drug development. New opportunities to accelerate drug discovery lie at the interface between machine learning methods, in this case developed for linear accelerators, and physics-based methods. The two in silico methods, each have their own advantages and limitations which, interestingly, complement each other. Here, we present an innovative method that combines both approaches to accelerate drug discovery. The scale of the resulting workflow is such that it is dependent on high performance computing. We have demonstrated the applicability of this workflow on four COVID-19 target proteins and our ability to perform the required large-scale calculations to identify lead compounds on a variety of supercomputers.